2005年度森基金 研究成果報告書
多種の網羅的生化学データを用いた
大腸菌中心炭素代謝経路の動的モデル構築
慶應義塾大学政策・メディア研究科
岩田 那由太
概要
大腸菌の中心炭素代謝経路は取り込んだ栄養分を分解してエネルギーを生成し,更に細胞を構成するための部品を供給する異化・同化の両性代謝の総称である.細胞内では産業的に有用な物質や薬剤として機能しうる酵素等が随時合成され続けており,これらの合成の多くにおいて中心炭素代謝経路が介在している.そのため微生物の改変を用いた有用物質の量産を考えた時,中心炭素代謝経路がどのように関与するのか,また,これをどのように改変すれば,目的とした結果が得られるのかを予測することは工学的にも生物学的にも非常に重要となってくる.しかしながら,細胞内では多数の因子が複雑に絡み合っているため,依然としてある一つの遺伝子の改変を施した時に細胞がどのような振る舞いをするのかを予測するのは困難である.
こうした背景の元,システム工学の力を用いて細胞の振る舞いを予測することができれば,有用物質生産プロセスを確立する際に様々なコストの削減につながる.ところが細胞シミュレーションに代表されるようなシステム工学的アプローチは,細胞内の実験データの不足によって頓挫することがあった.
そこで本研究では多種の網羅的生化学データ(マルチオミックスデータ)を用いて,ある環境にある細胞の定常状態近傍での振る舞いを予測できるようなモデルの構築及びその方法論の確立を行った.まずは希釈率0.2[hr-1]のオミックスデータを用いてパラメータチューニングを行った結果,当初の予定通り,物質の初期濃度の変動がない定常状態のモデルを作成することができた.次に希釈率0.1,
0.2, 0.5[hr-1]のオミックスデータを教師データとしてパラメータチューニングを行い,教師データとして利用していない希釈率0.4[hr-1]の定常状態物質濃度を予測したところ平均120%程度の誤差での予測に成功した.
*本誌は2005年度修士論文の短縮版である.
1. はじめに
産業革命以降全人口と局地的な人口密度の増大によって,農業の拡大,森林の剥奪,化石燃料の採掘と燃焼,環境汚染など,人間の活動は他の生物の分布と生長に著しく影響を及ぼしてきた[1].こうした時代にあり,地球規模での持続的な発展を維持し続けるために,21世紀は「化石資源依存型社会から生物資源依存型社会へ」大きなパラダイムの転換が求められている時代であるといわれており,微生物やバイオマスを利用した環境負荷の少ないプロセスの確立が求められている[2].
微生物の産業利用プロセスは試行錯誤と偶然の繰り返しによって生まれてきた.しかしながら,地球規模での環境循環調和社会の実現に向けて微生物を用いた産業応用は急務であると考えられるため,理論的なアプローチによる微生物の設計技術の確立が要求されている.分子生物学の発展に伴うDNA操作の発展,そしてオミックス解析とシステムバイオロジーを用いることで,微生物の遺伝子を操作した時に細胞内での挙動を予測し,物質生産を効率的に行うことのできる細胞を創生することが可能になりつつある.
そこで本研究では大腸菌を用いた中心炭素代謝経路のモデリングを行う.大腸菌は遺伝子数も約4000と比較的少なく,遺伝子工学の知見が豊富であるため,様々な変異や環境変化に対するオミックスデータを取得することが可能であり,微生物を用いた産業利用プロセスの基盤技術の開発に適していると考えられる.また中心炭素代謝経路は多くの研究が行われてきた系であるため,非構造化モデルのようにマクロな視点でのモデル化ではなく,個々の酵素反応に立脚したミクロな視点でのモデル化を行う経路として適している.
様々な物質が代謝される過程は,酵素がその触媒を行っているため,酵素濃度の変動が表現されること,すなわち遺伝子発現が正確に表現されている細胞モデルの構築は重要な課題である.しかしながら,こうした遺伝子発現を転写,翻訳,分解としてモデル化する体系化された手法は少ないため,より多くの研究がなされている代謝モデルに絞ったモデル化を検討する.将来遺伝子発現が細胞内のダイナミクスに組み込まれてくる実用的な段階に達した時に備え,本研究でのモデルの目的を以下のように設定し研究を行う.
「任意の定常状態近傍における酵素濃度が与えられた時に,任意の定常状態近傍の細胞内酵素活性を表現できるような大腸菌中心炭素代謝経路の動的モデルの構築」
2. 対象と方法
2-1. 大腸菌
本研究では対象とする生物として大腸菌を用いる.大腸菌はグラム陰性の通性嫌気性菌に属し,環境中に存在する主要な原核生物の一つである.遺伝子の総数は4000程度といわれており,ゲノムDNAは約470万bpからなる環状二本鎖である.バクテリアのモデル生物として様々な方面から研究がなされていて,とりわけ遺伝子工学的知見が豊富に蓄積されている[3].そのためある特定の遺伝子を欠損させた時の表現型を観察し,物質の流れを制御し有用な物質を量産するプロセスを確立するのに適した生物であると考えられる.
また大腸菌をコンピュータシミュレーションの対象生物とした時の利点として,連続培養による培養技術が確立されていることが挙げられる.連続培養ではすなわち菌体の比増殖速度や物質濃度が定常状態にあるとみなすことができ,その培養を瞬時に止めたデータを利用することで,ある定常状態にある細胞の各種情報を取得できる.
2-2. 中心炭素代謝経路
微生物が増殖する一連の流れは,グルコースを始めとする糖分の取り込み,その糖分を分解しATP*やNADPHなどの各種エネルギー通貨を生成する異化作用と,増殖に必要な様々な部品の前駆体および前駆体を元に各種アミノ酸,細胞壁成分,多糖類などを作る同化作用の二つに大別される.各種前駆体の数は,取り込み可能な栄養分の数および細胞増殖に必要な成分の数と比較して少ないため,細胞合成を行うための重要な制御点となる.異化作用から同化作用への一連のプロセスはそれぞれの段階で関与する要素の数の関係から蝶ネクタイ型といわれることがある[4].
中心炭素代謝経路とは前述したように,異化作用と前駆体合成の同化作用の総称で,大きく分けて解糖系,ペントースリン酸回路,クエン酸回路の三つのシステムによって構成されている.産業的に有用な物質や各種アミノ酸は中心炭素代謝経路によって作られる前駆体を出発点としているケースが多いため,中心炭素代謝経路に関わる様々な研究がなされてきた[5].以下の図は中心炭素代謝経路のパスウェイ図である.
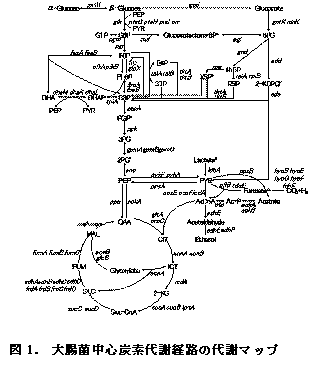
2-3. 反応速度論データ
モデル構築に必要なパラメータとして,吉野らの測定データ[6]および過去の文献の知見を用いた.使用したパラメータ,速度式など反応速度論情報は修士論文に記載した.以下に解糖系,ペントースリン酸回路,クエン酸回路の概略を示す.
・ 解糖系
*本文中で使用する略称対応表は修士論文に記載した。
解糖系は取り込まれたグルコースをピルビン酸まで分解し,2分子のATPを生成する.ほとんど全ての生物に共通に存在する糖の代謝経路で,別名Embden-Meyerhof経路とも呼ばれ,細胞質で反応が進行する.大腸菌の解糖系をモデル化した研究として[7]があり,本研究では解糖系の反応速度式は主にこのChassagnoleらの論文のものを参考にした.反応ごとの反応速度論パラメータは実験条件の違いや菌株,実験温度の違いなどを考慮して吉野らによって測定されたデータを利用した[6].
・ ペントースリン酸回路
ペントースリン酸回路はATP以外のもう一つの還元力エネルギー通貨であるNADPHを生成する酸化過程,核酸合成の前駆体となるリボース-5-リン酸を生成する非酸化過程の二つに分かれる.
・ クエン酸回路
クエン酸回路はHans Krebsによって提唱され,トリカルボン酸サイクル,TCAサイクル,Krebsサイクルなどと呼ばれる[8].クエン酸回路は糖,脂肪酸,アミノ酸を酸化的に代謝する経路であり,また同時に各種生合成前駆体の生成も担っている.好気条件ではアセチルCoAとオキサロ酢酸の縮合に始まり,最終的にオキサロ酢酸を再生するという環状型の反応系を有する.嫌気条件の時にも生合成に必要な各種前駆体を作るためにクエン酸回路は逆に回ることが知られている.
・ 細胞合成経路
モデルが全体として定常状態に達しているということはモデルの流入速度の総和と流出速度の総和が等しくならなければならない.本モデルでは炭素源であるグルコースを入り口とした時,細胞合成に必要な各種細胞構成部品を生成するための前駆体消費を出口経路としている.中心炭素代謝経路でグルコースが代謝されて行く過程で幾つかの物質は同化に必要な前駆体となる.前駆体となる物質は細胞の分裂要求に応じて細胞合成に必要な各種アミノ酸,多糖類,細胞壁などの材料へと変換されてゆく.個々の同化作用を正確にモデル化することは細胞シミュレーションの大きな課題であるが本研究の目的とは異なるため,前駆体を出発点として最終的に細胞構成成分が作られるまでの全ての反応を一つにまとめ,それを流出経路とみなした.
2-4. オミックスデータ
本研究ではこうしたオミックス解析によって取得された,各種網羅的な生化学データを用いて細胞モデルの構築を行った.モデルを構築するためにタンパク質濃度はショットガンプロテオミクスとウエスタンブロットを用いたプロテオミクス技術を,代謝産物濃度はCE-MSによるメタボロミクス技術を,細胞内酵素活性はGC-MSとNMRを用いたフラックソミクス技術を用いてデータの取得を行った.本研究で用いた培養およびオミックスデータの取得は慶應義塾大学先端生命科学研究所で行われた.まずこれらの個々の実験手法について概略を述べる.オミックスデータの議論およびモデルで使用したデータは修士論文に記載した.
・ プロテオーム
細胞内酵素濃度はショットガンプロテオミクスとウエスタンブロットよって定量が行われた.ウエスタンブロット法はニトロセルロース膜にサンプルのタンパク質をブロットし,ブロットされたタンパク質とそれに特異的な抗体との免疫反応によって目的のタンパク質を定量する方法である[9].タンパク質の定量方法としては歴史があり,1979年の[10]らの報告で既に使用されている.
ショットガンプロテオミクスは近年,慶應義塾大学先端生命科学研究所(IAB)で考案されたハイスループットなタンパク質定量方法である[11].まずスタンダードのタンパク質をLys-Cというプロテアーゼで切断し,その断片ペプチドをLS-MS/MSに導入する.すると一台目の分析器を通過した断片は衝突室でアルゴンなどの不活性化ガスと衝突解離されるので,得られるプロダクトイオンが二台目を通過した時に測定をする.これによって任意のタンパク質が衝突室の通過前後によって得られる断片ペプチドのプロファイルを取得することができる.次にサンプルを同様にプロテアーゼで切断し,LC-MS/MSに導入することでスタンダードと同様のプロファイルを見出し,サンプル内の目的タンパク質の定量化をすることができる.
・ メタボローム
代謝産物濃度はCE-TOF-MSによって測定されたメタボロームデータを用いた.CE-TOF-MSとはキャピラリー電気泳動法で物質の分離を行い,Time
of Flight(TOF)を使って質量対電荷比(m/z)で質量分離を行う測定装置である.測定結果は,コンピュータで処理され,マススペクトルなどの適当なデータとして出力される
[12].
・ フラックソーム
in vivoの定常状態における酵素活性(フラックス)としてはフラックス解析で得られた値を用いた.フラックス解析を行うには,まず炭素源として通常のグルコースの代わりに安定同位体である13Cでラベルされたグルコースを培地に添加する.そして最終的に菌体を加水分解し,GC-MSおよびNMRでアミノ酸の同位体分布を測定する.その結果得られた代謝物質の炭素原子の標識濃縮度,代謝物質の分子量分布,代謝物質のNMRスペクトルによって得られた強度の実験値と計算結果によって得られた値とを比較し,計算結果が実験値ともっとも近くなるようなフラックスを推定する[13].
2-5. オミックス,反応速度論データを用いたパラメータチューニング
本研究で構築するモデルは定常状態を実現するために,フラックス解析によって得られるフラックスに追従するような形でパラメータのチューニングを行った.例として,1基質1生成物で可逆型ミカエリスメンテン式の場合,以下に示す図のようになる.
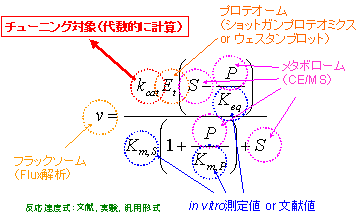
図 2. 一基質一生成物のミカエリスメンテン式とパラメータチューニング
ここで物質濃度はメタボロームの結果を,フラックスの値はフラックス解析の結果を,酵素濃度はプロテオームの結果を,KmやKeqは文献値およびin vitroの測定値を用いる.するとKcatのみが未知パラメータとなり,代数的にKcatを計算することができる.
Kcatは酵素反応速度実験の温度やpHに敏感であり,実験による誤差が大きいため,本研究ではKcatをチューニング対象として代数的にKcatを計算した.そしてそのチューニング結果をin
vitroの測定されたKcatと比較することで,チューニングの妥当性を検証した.
3. 結果・考察
3-1. 希釈率0.2[hr-1]におけるチューニング結果と考察
前述した方法によりチューニングを行い,チューニングパラメータの信頼性を把握するために,まず,希釈率0.2[hr-1]のオミックスデータを用いてin vitroで測定されたKcatとチューニングされたKcatとを比較した.
希釈率0.2[hr-1]の時のオミックスデータ,酵素反応速度論的パラメータは修士論文に記載した値を使用し,本誌では紙面の都合上割愛した.
表1. Kcatのチューニング結果と実測値の比較
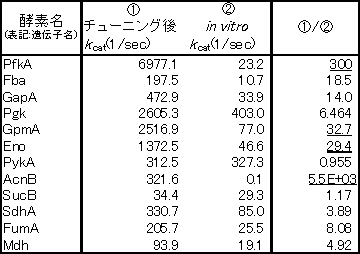
表1はチューニングしたKcatとin vitroで測定されたKcatとを比較したものである.そのPfkA,
GpmA, Eno, AcnBなどでチューニング結果と実測値との間に大きな開きがあることが分かった.PfkAは細胞内で調節酵素としての役割を担っており,様々な代謝物質による阻害や活性化によってPfkAの活性は制御されているため,現在モデルに含まれているPEP,
AMP, ADPのみでは反応速度式が正確に表現されていない可能性がある.AcnBではチューニングされたKcatの値は他の酵素と比較してあまり変わらないにも関わらず,in
vitroで測定されたKcatは他の酵素と比較して二桁のオーダで値が小さかった.過去のAcnBの酵素速度論文献を調べると[14]ではKcatは2.16となっており,本研究で用いた値の21.6倍も大きい.これらのことから酵素反応速度測定実験の過程でKcatが低く測定されてしまった可能性がある.
チューニングの結果,測定値とある程度一致するもの,全く一致しないものが得られた.総じてこれらの値が異なってくる理由として以下のような理由が考えられる.
1.
速度式,パラメータ,未知の影響物質が正確に考慮されていない可能性
2.
実験誤差
3.
in vivo とin vitroの違い
まず速度式の正確性についてだが,前述したように本モデルで使用している反応速度論パラメータは吉野らによって測定された実験値を用いている[6].吉野らはアロステリック酵素として知られているPykFやPfkAを除いて,他の酵素はミカエリスメンテン式に従うと仮定して試験を行った.このため,正確な反応機構に基づく反応速度式を用いていない場合がある.そして酵素は様々な阻害剤,活性剤のフィードバックによる影響を受けている.例えばPEPは解糖系だけで見てもGlk,
Pgi, PfkA, FbaA, PkFなどに影響を及ぼしている.既知の影響物質は酵素データベースのBrenda[15]や過去の文献により取得可能であるが,本モデルにおいて考慮していない因子がチューニング結果と実験値との違いを生んだ可能性がある.
次に考えられるのは実験の誤差である.生化学データは一般に正確な絶対値の測定が難しく,例えば本研究においてCE-Q-MSによって測定された陰イオンの中でモデル中に使用している物質の変動係数の平均は約30%(n=6)であった.同様にウエスタンブロットでは約39%であった.またフラックス解析における最適化計算の結果は使用した代謝経路に依存する.そのため,想定した経路が正確であれば問題ないが,細胞内の実際の経路が想定しているものと異なった場合には信頼性が低下する.
また,酵素反応速度論的パラメータについては,in vitro測定値はあくまで試験管の中で測定された値であり,in vivoの環境は試験管内と異なる可能性がある.例えばin vivoではin vitroと比較して総タンパク質濃度も多く時に細胞の30%にも達する[16].また[17]や[18]らは酵素同士の接近によって効率的な触媒を行うメタボライトチャネリングの存在などを指摘している.メタボライトチャンネリングによってin
vivoの酵素の活性がin vitroと比較して高くなっていれば,in vivo のデータから計算したKcatがin
vitroの測定値よりも概ね高くなっているということは合理的である可能性がある.そのため,こうしたin vivo とin
vitroの違いを考慮した上で値の評価をする必要性があるだろ.
このようにチューニング結果と実測値との間の大きな相違を生む原因は様々であるが,こうした問題点を把握した上でKcatだけでなくKmなどのほかのパラメータをもチューニング対象とし誤差の分散を図ることで,より真の値に近づく可能性がある.
3-2. 複数希釈率のオミックスデータに基づくパラメータチューニング
次に複数の希釈率を教師データとして用いたパラメータチューニングを行い,モデルの精度向上を図り,モデルの予測性について検討した.
連続培養では,希釈率が高くなると菌体濃度が0になるウォッシュアウトという現象がおきる.本研究の条件では,希釈率0.5[hr-1]まではウォッシュアウトがおきず安定に培養ができる(Data
not shown).そこで希釈率0.1〜0.5[hr-1]の範囲内にある定常状態を予測できるようなモデルの作成を試みた.
モデルの作成に当たって,本研究では慶應義塾大学先端生命科学研究所によって取得された希釈率0.1,0.2,0.4,0.5 [hr-1]のオミックスデータを用いる.0.7[hr-1]での連続培養も試みたが安定した培養ができなかった(Data
not shown).0.2[hr-1]のデータを使ったモデルは既に構築されており, 0.1[hr-1],0.5[hr-1]はそれぞれ使用可能な希釈率の下限と上限である.以上の理由からモデルを作成する際の教師データとしては0.1[hr-1],
0.2[hr-1], 0.5[hr-1]のデータを使用し,まずはそれぞれの希釈率を表現できるモデルを構築した.次に希釈率0.4[hr-1]の酵素濃度を入力した時の実験値と予想値とを比較することでモデルの予測性について検討する.
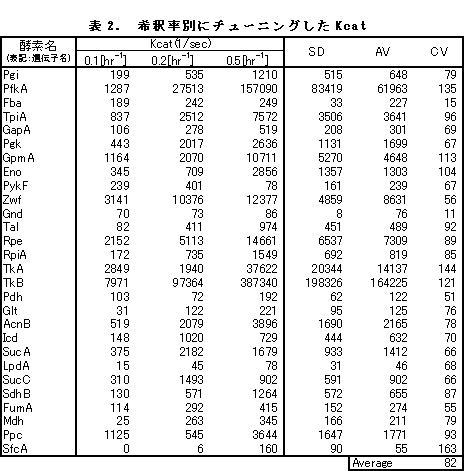
教師データとして使用するオミックスデータは修士論文に記載した値を使用し,本誌では割愛させて頂いた.チューニング方法は2-5で示した方法を用いて,それぞれの希釈率における,全ての酵素についてのKcatを計算した.表2はそれぞれの希釈率で計算したKcatの値である.チューニングしたKcatについての変動係数の全酵素の平均は約82%となった.個別の酵素ごとに見るとFbaやGndなどは変動係数が低く,逆にPfkA,GpmA,Eno,ペントースリン酸回路の酵素などの変動係数は高かった.
4. グルコース摂動実験による実験値とシミュレーションの比較
次にモデルの動的な振る舞いを確認するために,グルコースの過剰投与実験を行い,シミュレーションと実測値との定性的な振る舞いの比較を行った.グルコースの過剰投与実験は[7]らが定常状態時の30倍の濃度を投与した後の解糖系関連物質の濃度経時変化を測定しているため,その値を比較対象に用いた.ただし,シミュレーションで投与するグルコース量を30倍にしたところPtsの基質であるPEPが枯渇し計算が破綻してしまったため(Data
not shown),このような問題が生じない定常状態時の3倍の濃度を本シミュレーションにおけるグルコース投与量とした.[7]らは希釈率0.1[hr-1]でデータの取得を行っているので,本モデルも希釈率を0.1[hr-1]に変更し,希釈率0.1[hr-1]の時の酵素濃度測定値を入力して定常状態に達した時点でのモデルを利用した.
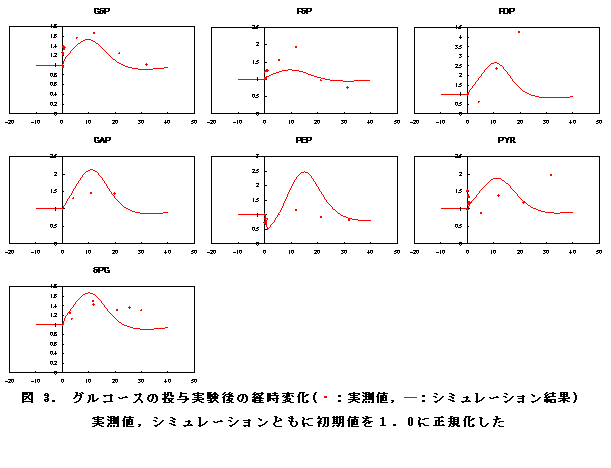
図3は0秒時点で任意のグルコース量を投与した後の解糖系関連物質の経時変化である.G6Pはグルコースを投与した後,シミュレーションと実験の両方で約10秒間蓄積され,その後約20秒をかけて元の定常状態に戻った.またPEPでもシミュレーションと実験の両方で挙動が一致しており,グルコースを投与した後に,約5秒間かけて濃度が低下し,その後再び濃度の上昇が見られた.これはグルコースを投与した後,PEPを基質として使用するPTSの活性が瞬間的に高まったことが原因であると考えられる.6PGはグルコース投与後の濃度上昇は一致した.しかしその後シミュレーションで6PGの濃度は元の定常状態に戻ったが,実験値では上昇したままである.
シミュレーションと実験の値が概ね一致したもの,部分的に一致したもの,全く一致しなかったものが得られた.このように細胞のダイナミックな挙動を正確に再現することができなかった理由がいくつか考えられる.
まず第一に考えられるのはモデルの精度であろう.既に3-2で示したようにPfkA,
GpmA, Eno, ペントースリン酸回路関連酵素のKcatの値は,とりわけ変動係数が高く値が一定しなかった.これらの値が一定しない原因のいくつかは3-1で示したように
1.
速度式,パラメータ,未知の影響物質が正確に考慮されていない可能性
2.
実験誤差
3.
in vivo とin vitroの違い
といったことが原因として考えられる.例えばGpmAは二基質二生成物の反応であり,現在モデルで使用しているミカエリスメンテン式を掛け合わせた速度式では表現しきれていない可能性がある.そこでこのような酵素が,複数基質を表現する際に使用されるOrdered
Bi-Bi反応やPing Pong Bi-Bi反応に従うと仮定し,これらの式に含まれる未知パラメータにフィッティングを行うといった試みが必要となるだろう.
そして第二にモデルで遺伝子発現を考慮していないことが考えられる.本モデルはある環境の定常状態近傍で酵素濃度が変化しない程度の環境の変化を保証することができる.そのため細胞の環境が劇的に変化し,実細胞では遺伝子発現の調節により環境の変動に対応していると考えられるような現象を再現できる保証がなくなってしまう.またChassagnoleらの実験条件ではグルコース濃度が30倍にまで高まった結果,呼吸による糖の消費がキャパシティを超え,醗酵経路が動くCrabtree効果が働く可能性がある[19].そして[20]ではグルコース濃度が変化することによって,外膜やペリプラズムのグルコース取り込みシステム自体が変化し,環境の変化に応答すると報告されている.このように細胞は環境の変化に応じて遺伝子発現による調節を行っているため,例えばグルコースの過剰投与のような環境の変化の細胞への影響を再現するためには,本モデルの個々の酵素を動的に表現する必要があるだろう.
5. 総括・展望
システムバイオロジーの目標の一つは,単細胞,多細胞を問わず進化の過程で細胞が生命活動を維持するために配置された分子同士の相互作用による,様々な環境への応答をあらゆるレベルで表現できるようになること,そして,その結果実験では困難な現象の理解に対してin silicoでの仮説を提唱できるようになることだろう.
本研究ではそうした最終目標に向けて,まずはモデル単細胞微生物である大腸菌の代謝の中で最も多く研究されてきた中心炭素代謝経路に焦点を絞りモデル化を行った.過去のモデルの多くで,要素の少ない系に絞ったモデル化が行われてきた理由の一つは実験データの不足であった.本研究では中心炭素代謝経路に関わるマルチオミックスデータと酵素反応速度論的パラメータを用いることで,実測値とシミュレーション結果の比率が平均120%に収まるモデルを作成することができた.また得られたパラメータの結果を用いて速度式,実験結果の信頼性,in vivo
とin vitroの相違という観点からモデルの精度を向上させるために留意すべきことを把握することができた.
パラメータチューニングの際に,まずはKcatをチューニングパラメータとしてモデルの開発を行った.今後はKcatだけでなく他のパラメータにも同様に誤差を分散させる手法が必要となってくるだろう.例えば本研究で使用した複数の希釈率におけるオミックスデータを用いることで,一つの酵素で解析的に解くことのできる未知パラメータの数を,希釈率の数にまで増やすことができる.そして解析的に求めるパラメータを選定するために,本研究で用いた手法を全てのパラメータに適用し,その結果得られた変動係数を指標にすることで,優先順位の高いものからチューニングするといった体系的なチューニング方法が可能になると考えられる.
細胞シミュレーションが真の力を発揮するのは実測値との結果が異なった時である.実験データを正しいとすると,相違が出る原因はモデルにあるはずである.例えば本研究で対象としているのは中心炭素代謝経路であるが,実際の細胞では想定範囲外の経路も動いているだろう.そこで想定していない経路の存在を実験的に確認し,それをモデルに加えることで実験データにより近づく可能性がある.また,本モデルではグルコース投与シミュレーションを行ったところ,グルコースの上昇に伴い,NADHが上昇し,電子伝達系が活性化され,ATPが上昇し,ADPが減少する(Data not shown).しかしながら,[7][21]では同様のグルコース投与実験が行われており,これらの報告ではグルコースの上昇に伴い,ATPが減少し,ADPが上昇するという本モデルでのシミュレーションとは逆の結果となっている.グルコースからG6Pへのリン酸基転移反応はPTSだけでなく,グルコキナーゼによっても進行する[3].このような相違が生じる原因の一つとして,シミュレーションではグルコキナーゼを考慮していないことが可能性として挙げられる.グルコキナーゼはグルコースへのリン酸基転移にPEPではなくATPを使用するため,過剰なグルコースの投与に伴ってPTSだけでなく,別の機構によってグルコースが取り込まれ,その結果グルコースを基質とするグルコキナーゼの活性が上昇し,ATPが減少しADPが上昇した可能性がある.
本手法ではある環境条件における酵素濃度は既知であるとしている.そのためモデルが保証するのはある環境条件での定常状態近傍である.今後遺伝子発現がモデル化されれば,環境条件の変動による酵素濃度も同時に計算することで,細胞の大きな環境の変化への適応も再現することができるようになるだろう.酵素自体の反応速度論的な特性は細胞外部の環境によって変化するものではないと考えられるため,様々な環境の酵素濃度を入力した時に,その環境近傍を正確に表現できるようになれば,遺伝子発現を含むモデルへの移行がスムーズに行われると考えられる.
6. 謝辞
本研究を進めていくにあたって慶應義塾大学・先端生命科学研究所の石井伸佳氏には研究の方針から細部まで多くの指導をしていただいた.この研究を形あるものにすることができたのも石井氏の力に拠るところは大きくこの場を借りて最大の感謝をしたいと思う.同・先端生命科学研究所の中山洋一氏からの科学者としての研究に対する姿勢からは学ぶことが大きく,本研究を進めてゆく上で常に立ち止まって非科学的になっていないかを確認することができた.同・先端生命科学研究所の菅原香織氏,五十嵐沙織氏にはCE-MSのデータを取得して頂き,平沢敬氏,那波幹氏,平井健太氏,には様々な希釈率での大腸菌の連続培養を実施して頂いた.同・先端生命科学研究所の富樫貴氏にはウェスタンブロットのデータを取得して頂いた.九州工業大学生命情報工学科のPei
Yee Ho氏にはそれぞれの希釈率におけるフラックス解析のデータを取得して頂いた.ヒューマンメタボロームテクノロジーズ株式会社の杉山直幸氏にはショットガンプロテオミックスのデータを取得して頂いた.愛知医科大学の吉野昌孝教授にはモデルを作成する際に使用した反応速度論的データを取得して頂いた.これらのデータなしに本研究を完成させることはできなかったため,これらの方々にはこの場を借りて感謝の意を述べたいと思う.慶應義塾大学政策・メディア研究科の戸谷吉博氏との研究に関する議論では得られることが非常に大きかった.同・総合政策学部の大薄麻未氏には修士論文執筆にあたる技術的な助力をして頂いた.同・先端生命科学研究所の柚木克之氏には研究室に所属してから4年間指導して頂いた.同・政策・メディア研究科中山研究グループ大学院生には研究から私生活まで様々な助言を頂き研究をする際のエネルギーを与えていただいた.そして,このような研究を行える環境を与えていただいた同・環境情報学部長兼同・先端生命科学研究所所長の冨田勝氏に感謝の意を述べたいと思う.
最後に生まれてから今に至るまであらゆる面で支えて頂いた家族に心より感謝を申し上げます.
本研究は新エネルギー産業技術総合開発機構(NEDO)「生物機能を活用した生産プロセスの基盤技術開発」の支援を受けて実施されました.
7. 参考文献
[1]スタニエ R. Y., et. al(1995)”微生物学 入門編”培風館, 東京.
[2]http://www.rish.kyoto-u.ac.jp/mission4.html
[3]Neidhart F. C., Curtiss R. (1996) “Escherichia coli and Salmonella 2nd
Ed.” Amer. Society for
Microbiology, Washington, D. C.
[4]Marie Csete, John Doyle, Bow ties, metabolism and disease, TRENDS in
biotechnology., 2004, 22(9).
[5]Stephanopoulos G., Metabolic fluxes and metabolic engineering,
Metab. Eng., 1999, 1, 1-11.
[6]Yoshino M., Non published data.
[7]Chassagnole C., Noisommit-Rizzi N., Schmid J. W., Mauch K., Reuss
M., Dynamic modeling of the central carbon metabolism of Escherichia coli,
Biotechnol. Bioeng., 2002, 79, 53-73.
[8]Krebs H. A., The tricarboxylic acid cycle, Harvey. Lect., 1948-1949,
44, 165-199.
[9]平野久(2001)”プロテオーム解析-理論と方法-”東京化学同人, 東京.
[10]Towbin H.,
Staehelin T., Gordon J., Electrophoretic transfer of proteins from
polyacrylamide gels to nitrocellulose sheets. Procedure and some applications,
Porc. Natl. Acad. Sci. U.S.A., 1979, 76, 4350-4354.
[11]Sugiyama N., Non published data.
[12]原田健一, 田口良, 橋本豊(2002)”生命科学のための最新マススペクトロメトリー ゲノム創薬をめざして”講談社サイエンティフィク, 東京.
[13]グレゴリ N ステファノポーラス
et. al.,(2002)”代謝工学 原理と方法論”東京電機大学出版局, 東京.
[14]Jordan A. P., Tang Y., Bradbury J. A., Thomson J. A., Guest R. J.,
Biochemical and spectroscopic characterization of Escherichia coli aconitases (AcnA and AcnB), Biochem. J., 1999,
344, 739-746.
[15]Schomburg I., Chang A., Schomburg D., BRENDA, enzyme data and
metabolic information, Nucleic Acids Res., 2002, 30(1), 47-49.
[16]Heijnen J. J., Approximative kinetic formats used in metabolic
network modeling, Biotechnol.
Bioeng., 2005, 91(5), 534-545.
[17]Cascante M., Franco R., Canela E. I., Use of implicit methods from
general sensitivity theory to develop a systematic approach to metabolic
control, I. Unbranched pathways. Math. Biosci., 1989, 94(2), 271-288.
[18]Vertessy B., Ovadi J., A simple approach to detect
active-site-directed enzyme-enzyme interactions. The
aldolase/glycerol-phosphate-dehydrogenase enzyme system, Eur. J. Biochem.,
1987, 164(3), 655-659.
[19]Thierie J., Modeling threshold phenomena, metabolic pathways
switches and signals in chemostat-cultivated cells: the Crabtree effect in
Saccharomyces cerevisiae, J. Theor. Biol., 2004, 226(4), 483-501.
[20]Ferenci T., Adaptation to life at micromolar nutrient levels: the
regulation of Escherichia coli glucose transport by endoincuction and cAMP,
FEMS. Microbiol. Rev., 1996, 18(4), 301-317.
[21]Aminul H. Md., Ushiyama H., Tomita M., Shimizu K., Dynamic responses
of the intracellular metabolite concentrations of the wild type and pykA mutant
Escherichia coli against pulse addition of glucose or NH3
under those limiting continuous cultures. Biochemical Engineering Journal,
2005, 26, 38-49.