Search of causative functional SNP using the in vitro virus method
Graduate School of Media and Governance
Bioinformatics Program
Hitomi Umeki
Abstract
Single Nucleotide Polymorphism (SNP) is the most common individual difference creating variety among individuals, and its relationship to disorders and therapeutic responses have been a concerning issue. Among the 10 million SNPs which exist in the human population, about 50,000 to 250,000 SNPs are thought to affect protein functions in vivo although there is still limited knowledge on the detailed mechanisms and their full identification.
Meanwhile, IVV method is a novel comprehensive, high-throughput protein-protein interaction (PPI) analysis method developed by Keio University's Dr.Yanagawa's research group. This method is unique in its accessibility to DNA information and capability of determining minimal PPI interacting regions.
Combining this IVV method with bioinformatics and public SNP database, NCBI's dbSNP (build 126), this work aims to explore the functional variety of proteins by searching for causative functional SNPs with alterations in their PPI.
With the establishment of a computational extraction system and the extraction of a nsSNP in c-jun as a major candidate, experimental verifications were performed to see whether it affected the PPI status with c-FOS. Subsequently, additional verifications were carried out through pull-down experiments of bait c-JUN and ETS1/MDM2/c-JUN. In addition, modifications in the candidate extraction system, such as the introduction of new software and characteristic approaches have allowed the selection of more accurate list of functional SNP candidates.
1. Introduction
With the completion of the Human Genome Project (HGP), and advances in comparative and functional genomics, the stream in molecular biology has shifted in gaining understandings of the human genome from multiple aspects such as function, structure, and diversity of genes among individuals. Single Nucleotide Polymorphism (SNP) is a mutation at a single nucleotide position and is the most common individual difference creating variety among individuals. With the establishment of public SNPs databases represented by NCBI's dbSNP, (Sherry et al., 2001 )and EBI's HGVbase (Fredman et al., 2004) combined with initiatives of International HapMap Project(Imyanitov et al., 2001)to create haplotype maps, the identification of SNPs linked to pathophysiology has become a critical challenge in this emerging field.
According to the Human Gene Mutation Database (Krawczak et al., 2000), missense mutations account for almost 50% of all DNA mutation know to cause genetic diseases whereas 70% of all missense mutations are reported neutral. Numerous attempts have been reported in identifying those "deleterious" mutations by determining the nature of the residue change, affects in structure, and sequential factors such as amino acid conservation.
The frequency of SNPs in the human genome is approximately once per 600-1000 bp, which totals to 10million within the human genome. Estimation reveals that 100,000 to 200,000 of them define a unique human genotype, and even less, 50,000 to 250,000 directly affect protein functions in vivo (Ching et al., 2002). Despite the growing interest in elucidating the relationship of SNPs to disorders and therapeutic responses, human genotype-phenotype relationships still remain complex and unclear. One approach towards understanding this phenomenon may be by considering the changes occurring in the Protein Protein Interaction (PPI) status of genes with the use of the in vitro virus method.
in vitro virus (IVV) method is a novel method developed by Keio University Department of Science and Technology's Dr.Yanagawa's research group, which links mRNAs to their encoded proteins through the attachment of puromycin to the 3'terminal end of the mRNA (Miyamoto-Sato, et al., 2005). Taking advantage of the nature of puromycins binding to the C-terminus of proteins, the cell-free co-translation of tagged bait and prey proteins enables the formation of protein complexes and an easy access to DNA information. The application of this method allows a comprehensive analysis of PPI, and the ability to detect the PPI of multiple preys against one bait protein allows a high-throughput analysis.
2.Method
The SNPs most likely to impact the protein function of a gene are a) coding region non-synonymous SNPs (nsSNPs) which alter the amino acid possibly affecting protein structure and b) SNPs that are located within a PPI domain where larger impact is anticipated. These SNPs are hypothesized to affect the protein functions as well as the PPI network. Thus those meeting these two qualifications were defined as candidates of functional SNPs in this work.
Through the combination of computational analysis and experimental verifications, these functional SNP were searched, verified and explored.
3.Results and Discussion
Extraction of functional SNP candidates
With the establishment of a computational system, analysis results from the 5th round of the 1st IVV selection plate performed in October 2005 demonstrated c-jun as a candidate gene brought for experimetal verification.
The nsSNP of c-jun (rs9989) is yet not validated without a known phenotype. However, detailed survey and analysis revealed it to be a potential candidate of a functional SNP. Firstly, this Thr297Met nsSNP was found only among ETS1 sequences of adenocarcinoma pointing to a possible relationship with the disease (Buetow et al., 1999). Secondly, it is included within the bZIP and leucine zipper domain area of c-jun where studies have reported a high conservation rate among the JUN family compared to other areas (Schaefer et al., 2001). Numerous studies have indicated nsSNP found in highly conserved areas to create a larger impact on the protein function. Thirdly, c-JUN is well known for its strong PPI with c-FOS which is highly detectable through the pull-down method. Both are transcription factors and proto-oncogenes actively involved in events of cell differentiation, transformation, proliferation, and apoptosis. The two proteins heterodimerize and form an activator protein (AP)-1 family complex, to bind with DNA regulatory sequences (Janssen et al., 1997). Finally, despite being subjected to study for years, no other nsSNPs have been reported in the genes of c-fos or c-jun according to dbSNP.
Given these reports and evidence, it was hypothesized that this particular nsSNP may produce an impact on its protein structure and function. This hypothesis is supported by the results from Ramenskey et al.'s software PolyPhen (http://www.bork.embl-heidelberg.de/PolyPhen), which predicted the impact of the nsSNPs on a three-dimensional scale, scoring the mutation as "probably damaging". The positional relationships of c-jun and nsSNP are show below in Figure 1.
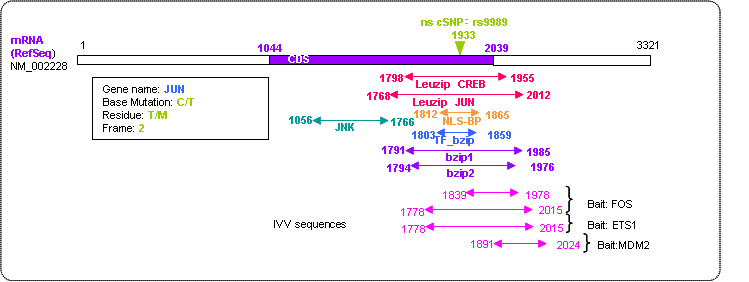
Figure 1 structure of c-jun
Experimental Verification of rs9989 nsSNP on c-JUN in its interaction with prey FOS, ETS1, MDM2 and c-JUN
In exploring the impact of the rs9989 nsSNP on its coding c-JUN protein function, pull-down assays with prey protein FOS, ETS1, MDM2, and JUN(homodimer) were carried out.
c-JUN and FOS are known for their strong PPI interactions with each other. Both are transcription factor proto-oncogenes and are active in events of cell differentiation, transformation, proliferation, and apoptosis. The two proteins heterodimerize and form an activator protein (AP)-1 family complex, to bind with DNA regulatory sequences (Janssen et. al. 1997). Studies have shown proteins in the JUN family to contain identical amino acid sequences in the DNA-binding bZIP domains while not in other areas (Schaefer et. al. 2001). The nsSNP verified in this work was included within the b-zip and Leucine zipper domain area of c-jun.
Functional cooperation of ETS and AP-1 is crucial for the controlled expression of many genes (Gottschalk et al., 1993), while studies have reported a nearby mutation of rs9989 in c-JUN creates a dynamic impact on the interaction with NFAT family protein, which ETS1 is a member of. Therefore differences between the wild type and mutant bait c-JUN were highly expected in the PPI with prey ETS1.
However, no difference in PPI with all preys were observed from the pull-down assays. A likely explanation is the insignificance of this nsSNP in protein conformation. T297M mutation resulted in the change of polarity from non-polar to polar, but remained as a neutral amino acid, also surrounded by neutral amino acids.
Meanwhile, studies report the Ser243Phe mutation of c-JUN to eliminate abilities as a -425rPRL promoter inhibitor, obtaining a function similar to v-JUN. This is caused by the loss of serine as a negative regulatory phosphorylation site (Bohman et al., 1987). Phosphorylation involves an addition of a phosphate group to a protein, resulting in a functional change of the protein in PPI, enzyme activity and cellular location. The phenomenon is reported to affect tyrosines, serines, and threonines, and the Thr297Met of c-JUN may result in the loss of a phosphorylation site. Along with the SNP analysis, combining a phospholyration analysis using databases such as Phosphobase (http://phospho.elm.eu.org/) may become relevant in future work.
4.Conclusion
The aim of this work was to search for functional and causative SNPs, affecting protein structure, function and PPI networks. In an attempt to reach this goal, candidate nsSNPs including genes were selected for verification through pull-down experiments. The one and only registered nsSNP of c-jun, rs9989, was predicted to produce a damaging impact supported by various evidence and predictions. Pull-down assays were carried out to determine changes in the PPI status between c-JUN and the mutant c-JUN-snp, but difference in PPI with c-FOS and ETS1 were not observed. The analysis will be continued with the conduction of the pull-down assay with MDM2 as prey proteins. In response to the updates of data and, new candidates were extracted and reconsidered, along with a characteristic analysis of diseases relating SNPs. In this work, the analysis of baits along with preys was newly applied, and a characteristical analysis found arginine to be involved in about 30%disease-related mutations registered in OMIM. Additionally, the consideration of phospholyration sites was proposed for future work. With the use of software such as PolyPhen, and numerous mutation databases, candidate nsSNP were further narrowed.
5.Prospective
The following approaches may be relevant for future works. 1) pull-down assays of bait c-JUN with prey MDM2. 2) in vitro virus experiment of c-JUN and c-JUN-snp to see whether there is a difference in the attracted prey proteins. 3) construction of TPI1 gene and conducting a in vitro virus experiment. Despite the IVV prey sequences and the TPI1 gene not carrying the same reading frame, numerous reports indicate its relationship with diseases and protein instability. Therefore, although pull-down experiment with the detected bait may not be informational, interaction differences are highly anticipated as it attracts different preys through the in vitro virus. 4) Construction of the new candidate gene and verifying whether they may show difference in its interaction with bait proteins through pull-down assays. 5) Characteristic analysis and consideration of phospholyration sites in extracting strong functional and causative nsSNP candidates.
6.Acknowledgements
Part of this work was supported by grants of the Genome Network Project from the Ministry of Education, Culture, Sports, Science and Technology, Japan. The quality of this experiment was greatly enhanced by the generous support of Professor Yanagawa and data providence of Keio University Faculty of Science and Technology, Yanagawa Lab. Assistant Professor Etsuko Miyamoto-Sato and Takanori Washio were extremely helpful in guiding this work with their valuable insights and advices. My deep gratitude goes to Dr. Masamichi Ishizaka and Dr. Shigeo Fujimori for continuous experimental and technical support. Discussions with Naohiro Yanagisawa., Noriyuki Kitagawa were invaluable as well as the help of all members' of the Yanagawa and Tomita Lab. Last, but definitely not least, I would like to thank Professor Masaru Tomita for this special opportunity and inspiration.
7.Special notes on the classified IVV data of Genome Network
This project is in part of the Genome Network Project(GNP) of the Ministry of Education, Culture, Sports, Science and Technology, and according to the regulations of "Genome Network Consortium implementation," the publication of all detailed results are forbidden. Because of these circumstances, the distribution of this work outside Keio University is strictly forbidden. We request that well care is taken towards the handling of data.
References
Alber, T., and Kowasaki G., (1982). J. Mol Appl. Genet. 1, 419-434.
Bohman, D., Bos, T., Admon, A., Nishimura, T., Vogt, PK, Tjian R. (1987) Science. 238. 1386-1392.
Bosselut, R., Levin, J., Adjadj, E., and Ghysdael J. (1993) Nucleic Acids Res. 21(22). 5184-5191.
Buetow, K.H., Edmonson M.N. and Cassidy A.B. (1999) Nature Genetics, 21, 323-235.
Ching, A., Caldwell, K.S., Jung, M., Dolan, M., Smith, O.S., Tingey, S., Morgante, M., and Rafalski, A.J. (2002). BMC Genet. 8, 19-33.
Cavallo A, Martin AC. (2005) Bioinformatics. 21,1443-50.
Coma M., Guix F. X., Uribesalgo I., Espuna G., Sol? M., Andreu D., Munoz F. J. (2005) Brain.128, 1613-1621.
Day SM., Westwall MV., Fomicheva EV., Hoyer K., Yasuda S., Cross NVL., Alecy LGD., Ingwall JS. And Metzger JM. Nature Medicine, 12, 181-189.
Daar IO.., Artymiuk PJ., Phillips DC. and Maquat LE. (1987) PNAS, 83,7903-7907.
Fredman, D., Munns, G., Rios, D., Sjoholm, F., Siegfried, M., Lenhard, B., Lehvaslaiho, H., and Brookes A. J. (2004) Nucleic Acids Res., 32, D516-D519.
Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Nucleic Acids Res. 2005;33:D514-7.Higgins D, Thompson J, Gibson T, Thompson JD, Higgins DG, Gibson TJ (1994) Nucleic Acids Res. 22,4673-4680.
Gottschalk LR, Giannola DM and Emerson SG. (1993). J. Exp. Med. 178, 1681-1692.
Imyanitov, EN., Togo, AV. and Hanson KP. (2004) Cancer Le..t, 204, 3-14.
Janssen YM, Heintz NH, Marsh JP, Borm PJ and Mossman BT. (1994) Am. J. Respir. Cell Mol. Biol., 11, 522-530.
Krawczak M, Ball EV, Fenton I, Stenson PD, Abeysinghe S, Thomas N, Cooper DN (2000) Human Mutation 15(1), 45-51.
Mande SC, Mainfroid VK., Kalk KH., Martial JA. And Hol, WGJ. (1994) Protein Sci. 3, 810-821.
Miyamoto-Sato, E., Ishizaka, M., Horisawa, K., Takashima, H., Fuse, S., Sue, K,. Hirai, N., Masuoka, K. and Yanagawa,
H., (2005) Genome Res, 15, 710-7.
Miyamoto-Sato, E., Takashima, H., Fuse, S., Sue, K,. Ishizaka, M., Tateyama, S., Horisawa, K., Sawasaki, T., Endo, Y. and Yanagawa, H., (2003) Genome Res, 15, 710-7.
Olah J., Orosz F., Keseru GM., Kovari Z., Kovacs J., Hollan S. and Ovadi J. (2002) Biochemical Society Transactions, 30, 30-33.
Olah J., Orosz F., Puskas LG., Laszio HJ., Horany M., Polgar L., Hollan S. and Ovadi J. (2005) Biochem J. 392, 675-683.
Pearson WR., Lipman DJ. (1988) PNAS, 85, 2444-2448.
Peterson BR., Sun LJ and Verdine GJ., (1996) PNAS, 93, 13671-13676.
Pham, C. G.; Bubici, C.; Zazzeroni, F.; Papa, S.; Jones, J.; Alvarez, K.; Jayawardena, S.; De Smaele, E.; Cong, R.; Beaumont, C.; Torti, F. M.; Torti, S. V.; Franzoso, G. (2004)Cell 119, 529-542.
Pichersky, EL., Gottlieb D., and Hess JF. (1984) Mol. Gen. Genet. 195, 314-320.
Pruitt KD., Tatusova T, Maglott DR. (2005) Nucleic Acids Res. 33(Database issue), D501-4.
Ramensky, V., Bork, P. and Sunyaev, S. (2002) Nucleic Acids Res., 30, 3894-3900.[
Schaefer LK., Wang S., and Schaefer, T. (2001) J. Biol. Chem., 276, 43074-43082.
Sherry, ST., Ward, MH., Kholodov, MB., Phan, L., Smigieiski, EM. And Sirotkin, K. (2001) Nucleic Acids Res, 29, 308-311.
Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, Abeysinghe S, Krawczak M, Cooper DN. (2003) Hum Mutat. 6, 577-81.
Stevens, T. H.; Forgac, M. (1997). Annu. Rev. Cell Dev. Biol. 13, 779-808, 1997.
Wang, Z. and Moult J. (2001) Human Mutation, 17, 263-271.