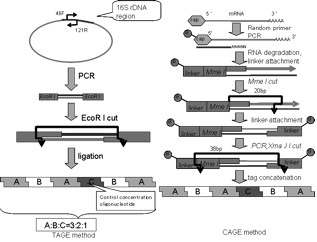
Figure 1. TAGE法とCAGE法の比較
慶応義塾大学 政策メディア研究科 修士課程1年 バイオインフォマティクス
奈須野 恵理
身の回りの環境における微生物群集と環境の状態を把握するためにモニタリングが重要な意味を持つと言える.モニタリングを行う上で試料中の微生物の定性だけでなく定量解析を同時に行うことが重要であるが,現在メタゲノム解析手法として主流となっている変性剤濃度勾配ゲル電気泳動法(Denaturing Gradient Gel Electrophoresis;DGGE)は定性解析に優れていても同時に定量性がない.そこで,メタゲノム解析プロジェクトでは転写発現解析で広く用いられているCAGE(cap analysis gene expression)法のストラテジーをメタゲノム解析に応用することで定性と定量解析を同時に,かつ他のメタゲノム解析手法と比べてより簡便な操作と装置で可能にする新規TAGE(Tag-Attached Gene Extention)法を考案し,2006年度より手法構築に向けた条件検討を行ってきた.その結果,TAGE法を用いることで得られたシーケンスデータから複数の生物種由来のDNA配列を1度に同定し,さらに試料中の種ごと分布比較が可能であることが実証された.
新規手法構築における条件検討が必要な項目として,全細菌に保存されている16S rRNA遺伝子領域においていかに短く,かつ種の分類が可能な領域を増幅するプライマーセットを用いてDNA断片をPCRで増幅するか,それらをライゲーションによっていかに多く繋げるか,そしてそれをプラスミドに挿入し一定以上の断片数を保持したクローンをいかに得るか,シーケンスでいかに多く読み取って各断片を定性・定量できるかが挙げられる.先学期までにプライマーセットとPCR条件を決定し,ライゲーション効率を上げる方法を検討した.今学期はベクターとインサートのライゲーション方法とクローニング効率の関係を明確にし,一定の個数以上のインサートを持つプラスミドを最も効率よく得られる条件を検討したが,挿入インサート数の平均を上げる条件を決定するに至らなかった.本報告書では考え得る操作上の問題を列挙し検証した結果の考察と共に,今後さらに詳細に検討すべき条件についてまとめる.
Key words: Metagenome, 16S rRNA, tandemrepeat ligation
こうして得たライゲーション産物を1つのインサートとしてベクターに挿入する.それをクローニングして得られるプラスミドを抽出しシーケンス解析することで1つのプラスミドから同時に複数の細菌種を同定できる(Fig.2).
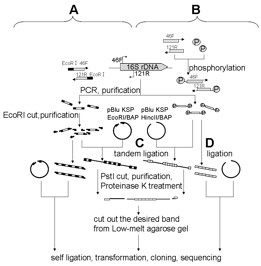
2007年度を通して、ライゲーションとクローニングの条件検討をする目的で約100bpの目的断片に加えて3’側に長い約500bpの断片を増幅する2種類のプライマーセットを用いてそれぞれPCRを行い,制限酵素処理と精製を行ったインサート断片とベクターを2通りのライゲーション方法(対象と手法参照)によって環状化したプラスミドを作製した.それをトランスフォーメーションして得られたクローンのプラスミドをシーケンスすることでクローニングサイトに挿入された目的断片の個数と種類を同定した.しかし,検討する条件が多く条件と結果の関連性が明確でなかったため,実験の操作上最も容易で時間短縮できるという理由から平滑末端断片のインサートに限定して年度後半は条件検討を行った.
本手法の元となっているCAGE法では各転写物を識別するために20bp前後の断片を対象としており,これは配列特異性を保ちつつ一度にシーケンス可能な500bp前後の長さの中に詰め込む配列の数を最大にした結果である.TAGE法の開発においてもこれにならってなるべく接近したプライマーセットのデザインを試みた.一般的にPCRやシーケンスを介する真正細菌のメタゲノム解析には全菌種が有していて他の分子に比べて保存性が非常に高く,データベースも充実しているなどの理由から16S rDNAが用いられる.そこでTAGE法においてもPCR増幅対象領域として採用した.約100bpを増幅する46F-121Rオリジナルプライマーに加えて,インサートの個数によるライゲーション断片の長さの違いがわかりやすい約500bpを増幅する46F-531Rプライマーも用意した(Tab.1).さらに,約100bpのインサート断片が長く繋がったプラスミドを持つクローンがこれまでの実験で得られなかったため,末端の配列がライゲーション効率を著しく低下させている可能性を考えてユニバーサル5Fプライマーと121Rの組み合わせも今学期使用した. ただし16S rRNAは上記の通り保存性の高い領域であり,その中でも種特異性の高い領域を選択する必要がある[6].46F-121RプライマーのTm値はどちらも適正範囲にあり,計算上全ゲノム解読済みの細菌420種のうち356種はこのプライマーで増幅した70bp前後の配列の違いによって種を区別することができると予想される(Tab.1).
インサートとベクターを混ぜるタイミングを2通り検証し,ライゲーション効率に差が現れるかを調べた.
一つ目の方法は始めにリン酸付き平滑末端インサート断片のみで1時間タンデムにライゲーションさせ,精製後に再度低温で脱リン酸化ベクターと混ぜてセルフライゲーションさせて環状プラスミドを得た.しかしインサートのみのライゲーションが高効率で起こった場合に生じるベクター以上の長い断片はクローニング後に得られないため,適度な長さにインサートが繋がった断片が多く生成されることが望ましい.
そこで,二つ目はインサート断片と脱リン酸化したベクターの分子数の比率を変えて混ぜたサンプルを用意して37℃で1時間ライゲーションさせ,インサートだけで長く繋がり過ぎることを防ぐと同時に両者の混合比率を変えることでインサート連結数をコントロールすることを試みた.ライゲーション後のサンプルに制限酵素用Hバッファーを溶液の1/5容加えて65℃で15分温めた.Hバッファーを加えることでサンプルの塩濃度を上げてDNAの二重鎖の変性を防ぐと同時にLigaseを失活させ,次の制限酵素処理中にライゲーション反応が起こらないようにした.そこへベクターを一カ所切断するPstIを加えて37℃で1時間制限酵素処理を行った.
両方法共に低温でのセルフライゲーション前の精製ではDNA断片に結合しているLigaseをはがすためにPromega社のProteinase K溶液(0.2% SDSとその約1/60容Proteinase K)を加えて50℃で1時間インキュベーションした.精製は一般的なフェノール,クロロホルム処理とエタノール沈殿によって行った.16℃でのセルフライゲーションにはTaKaRa社のMighty cloning Kit(Blunt end)に添付されているMighty mix試薬を用いて1時間以上反応を行った.
既存の微生物同定用DNAデータベースと系統解析ソフトの例としてテクノスルガ・ラボと国立遺伝学研究所が共同開発した「アポロン」が挙げられる.このデータベースは国際塩基配列データベースに登録されている細菌・放線菌・古細菌の既知5,247種(2006現在)におけるリボソームDNA塩基配列情報を網羅している.機能としてはシーケンスデータをデータベース内でBlast検索した結果を視覚的に見やすく表示し,マルチプルアライメントの結果や系統樹の出力が可能である.しかしこのソフト自体が420,000円と高額であるだけでなく,データベースの年間ライセンスが細菌だけで420,000円,細菌と真菌を合わせると840,000円要する.機能性と信頼性は高くても今後メタゲノム解析分野の発展を考える上では多くの研究所で手軽に利用できる金額ではない.
そこで,NCBIの管理しているデータベースを利用して独自のツールを開発した.まず,全既知細菌の16S rDNA配列情報を入手し,46F-121Rプライマーで増幅する領域のみを抽出して新たにデータベースを構築した.ファスタ形式のシーケンス結果から,オリジナル16S rDNAデータベースをBlast検索して各インサート断片の候補細菌名を絞った.次に既知細菌名の総合データベースに照らし合わせ,さらに微生物全般に関する界,門,綱,目,科,属,種の系統分類学的情報データベースに照らし合わせることで系統をさかのぼった.
まず50℃と55℃のアニーリングの温度と生物種によるDNA配列の違い,121Rと531Rプライマーを用いた場合のPCR増幅効率の違いを検証した(Fig. 3).この結果から,50℃と55℃でのPCR産物に有意な差は見られなかった.一般的にアニーリングの温度が低いほうが非特異的な産物が増えやすいといわれているが,この場合は50℃であっても特に増えていない.ただしリン酸化した46F-121RプライマーのPCR産物にはどちらの温度でも非特異的な産物が増えていることがわかる.また,PCR産物の長さに関係なくリン酸化プライマーよりEco RIサイト付きプライマーを用いた反応の方が増幅効率が若干良いという結果が得られた.
生物種ごとのPCR増幅効率はどのPCR産物でも大腸菌と枯草菌で比較すると若干大腸菌の方が良いと言える.
インサートを混ぜる比率が下がるとライゲーション後であるにも関わらずベクター1本分と2本繋がった分の長さのバンドがより濃くなった(Fig.4 lane2-4).このライゲーションサンプルをクローニングサイトであるHincIIの前後であるBamHIとPstIでダブルダイゼッションすれば挿入インサート部分がベクター以外の部分として細かく分離すると予想したが,実際はどのサンプルもベクターのみを現すバンドが明確になっただけでそれ以外は明確なバンドが観察できなかった(Fig.4 lane5-7).クローニング結果に差が出るかどうかを調べるためにベクター内をHindIIIで一箇所切断したサンプルも泳動で確認したが,バンドとして確認できるものはほぼインサートが入っていなかった(Fig.4 lane8-10).
クローニングの結果として、インサートの量が多いほどインサートゼロのクローンの割合が少なかった.ベクターに挿入した外来遺伝子が構造的に不安定であったり宿主となる大腸菌にとって有害であるタンパク質をコードしていたりする場合は複製のたびに挿入断片が削れて行く現象が知られている[8,9].一度短いプラスミドが生じると複製される時間がより短くなるため,コロニーを形成するほど細胞分裂した宿主内のプラスミドは削られて短いインサートを持つものが優勢になると考えられる.そして外来遺伝子の安定性はベクターのコピー数に反比例するため,短い繰り返し配列のような構造的に不安定な挿入断片をクローニングする際はプラスミドのコピー数が少ないものを選択する方が良いと予想される.また、マルチクローニングサイトの保存性を調べるために得られたコロニーからインサートが比較的長く挿入されているクローンと,主な太いバンドが見られない異質なクローンを5つ選別して液体培養し,プラスミドを抽出した.それらをクローニングサイトの外側2箇所で切断するPvuIIと,BamHI・HindIIIでそれぞれ消化してベクターから分離するバンドの長さを確かめた.
この結果から,コロニーPCR後の泳動で長い位置に太いバンドが存在するクローンは複数のインサートを持っている(Fig.5 A.lane1,3)のに対して非特異的産物しか見られないクローンはインサートゼロ(Fig.5 A.lane2,4,5)であることがわかった.マルチクローニングサイトの外側で切断されるPvuIIでは全てのプラスミドで切断されてバンドが2カ所ずつ見えるのに対して,BamHIとHindIIIで消化した際は切れ残りや全く切れていないものばかりであった(Fig.5 A.lane6-10).そこで,1~5のプラスミドをそれぞれBamHIとHindIII単独で切断して切断効率に差が生じるかどうかを調べたところ,インサートが複数挿入されている1と3のプラスミドはBamHI,HindIII共に切断されてリニアのバンドが見られる(Fig.5 B.lane6,11と8,13)のに対し,2と5はHindIIIでのみ切断され(Fig.5 B.lane7,12と10,15),4はどちらも切断されなかった(Fig.5 B.lane9,14).一連の制限酵素処理の結果から,抽出したプラスミドのクローニングサイトは正確に保存されておらず,HincIIに近いサイトほどライゲーションの際に削れている場合が多いことがわかった.このことから,インサートとベクターをライゲーションさせる時点でクローニングサイト周辺の塩基配列が壊れているために目的断片がベクターに正確に挿入されにくい可能性も考えられる.
繰り返し配列の構造的不安定さとマルチクローニングサイトの保存性が低い点がクローニング効率を著しく下げている可能性が高い。今後は再度使用するバッファーや水、試薬類などにDNaseがコンタミしていないかを確認し、さらに来年度はコピー数の少ないベクターを用いて同様の手法を行う予定である。さらに,今回開発した微生物同定用オリジナル16S rDNAデータベースと系統解析ツールをより正確に,かつユーザーの任意で出力結果を柔軟に変えられるように改良する.また,系統樹の描画やどの属由来のフラグメントがサンプル内にそれぞれ何割ずつ含まれるのかの結果を視覚的にわかりやすく表示する機能も追加することで今後扱う複雑な細菌群集サンプルの解析を迅速にわかりやすくする.
Eri Nasuno(Feb. 29th, 2008)