SVMを用いたヌクレオソームポジショニング予測
政策・メディア研究科 修士一年
小川 隆
小川 隆
近年、シーケンシングの発達やomics研究、及びゲノムワイドな実験によって様々なデータが蓄積され、新たな発見が数多く見つかっているものの、発現機構が解明されているとは言い難い。真確生物の発現システムにおける重要な機構としてヌクレオソームがある。ヌクレオソームはDNAが染色体を形成するために必要な構造で、発現制御に大きく関わっている。最近の研究でヌクレオソームポジショニングをゲノムワイドに取得する試みが精力的に行われている。本研究ではSaccharomyces cerevisiae(S. cerevisiae)においてChIP-chip法で得られたヌクレオソームポジショニングデータからSupport Vector Machine(SVM)による機械学習によって配列特徴を抽出し、予測を行った。SVMを用いたヌクレオソームポジショニング予測手法としてPeckhamらの手法があげられるが、学習データの抽出法の違いにより、本手法は高い予測精度を得た。
本予測手法によって得られたヌクレオソームポジショニング配列を、ChIP-chip法によって実験的に得られた配列との特徴比較を行った。配列特徴はAT/GC contentとDNA柔軟性において比較を行った。AT/GC contentでは予測配列と実験配列に同じ傾向が見られたがDNA柔軟性については見られなかった。 ヌクレオソームポジショニングを高精度で予測する本手法は、発現機構の解明に貢献することが期待される。
キーワード:
ヌクレオソームポジショニング, Support Vector Machine, 予測, DNA高次構造
本予測手法によって得られたヌクレオソームポジショニング配列を、ChIP-chip法によって実験的に得られた配列との特徴比較を行った。配列特徴はAT/GC contentとDNA柔軟性において比較を行った。AT/GC contentでは予測配列と実験配列に同じ傾向が見られたがDNA柔軟性については見られなかった。 ヌクレオソームポジショニングを高精度で予測する本手法は、発現機構の解明に貢献することが期待される。
キーワード:
ヌクレオソームポジショニング, Support Vector Machine, 予測, DNA高次構造
1.1 ヌクレオソーム
近年,シーケンサーの発達やomics研究により,生命というシステムを解明するための鍵となる膨大な量のデータが得られてきている.しかし,生命の機構は未だ解明されたとは言い難い.生命活動に必要なタンパク質,RNAといった分子を生み出す発現機構においても新たな知見が次々に得られている段階である.
発現機構における最近の大きな発見の一つとして,ヌクレオソームポジショニングが挙げられる.ヌクレオソームとは,ヒストンと呼ばれる樽型タンパク質にDNAが約1.65周,約150bp巻き付いた構造のことである.DNAは,ヌクレオソームを形成する領域と,リンカーと呼ばれる約50bpのヌクレオソームを形成しない領域が交互に出現し,電子顕微鏡で観察した時に”糸を通したビーズ”のように見える構造をとる.ヌクレオソームがさらに幾何学的に整列されることによって染色体を形成し,核内にDNAをコンパクトに収納するだけでなく厳密な発現制御にも関わっていると言われている.ヌクレオソームを形成しているDNAは他のタンパク質の接近が困難になる.そのため,ヌクレオソーム構造とヒストンから乖離したヌクレオソームを形成しない構造とのダイナミックな移行は,厳密な発現制御において重要な役割を持っている[1-4].これを見てもわかる通り,ヌクレオソームは生命にとって非常に重要なエピジェネティック機構であるといえる.
ヌクレオソームの形成する位置をヌクレオソームポジショニングと呼ぶ.最近まではヌクレオソームはDNA上に自由に結合すると考えられてきたが,DNAの配列によって親和性は異なり,定まった位置に形成されるヌクレオソームが多数あることがS. cerevisiaeにおいて実験的に発見された.ヌクレオソームが発現と深く関わっているとされるだけに,ヌクレオソームポジショニングは,発現機構を解明する大きな要素として注目を集めている[5-9].
発現機構における最近の大きな発見の一つとして,ヌクレオソームポジショニングが挙げられる.ヌクレオソームとは,ヒストンと呼ばれる樽型タンパク質にDNAが約1.65周,約150bp巻き付いた構造のことである.DNAは,ヌクレオソームを形成する領域と,リンカーと呼ばれる約50bpのヌクレオソームを形成しない領域が交互に出現し,電子顕微鏡で観察した時に”糸を通したビーズ”のように見える構造をとる.ヌクレオソームがさらに幾何学的に整列されることによって染色体を形成し,核内にDNAをコンパクトに収納するだけでなく厳密な発現制御にも関わっていると言われている.ヌクレオソームを形成しているDNAは他のタンパク質の接近が困難になる.そのため,ヌクレオソーム構造とヒストンから乖離したヌクレオソームを形成しない構造とのダイナミックな移行は,厳密な発現制御において重要な役割を持っている[1-4].これを見てもわかる通り,ヌクレオソームは生命にとって非常に重要なエピジェネティック機構であるといえる.
ヌクレオソームの形成する位置をヌクレオソームポジショニングと呼ぶ.最近まではヌクレオソームはDNA上に自由に結合すると考えられてきたが,DNAの配列によって親和性は異なり,定まった位置に形成されるヌクレオソームが多数あることがS. cerevisiaeにおいて実験的に発見された.ヌクレオソームが発現と深く関わっているとされるだけに,ヌクレオソームポジショニングは,発現機構を解明する大きな要素として注目を集めている[5-9].
1.2 ヌクレオソームポジショニング
近年,ヌクレオソームを形成するDNA配列の大規模な実験的取得による,ヌクレオソームポジショニングを解明する研究が精力的に行われている.クロマチン免疫沈降法(ChIP法)を用いて,ヌクレオソームをDNAに固定し,リンカー部分をDNaseによって分解する.これによって,ヌクレオソームを形成するDNA配列だけが残る.この残った配列は,タイリングアレイで取得する方法と,シーケンシングによって取得する方法とがあり,それぞれChIP法を含めてChIP-chip法,ChIp-seq法と呼ばれている.これらの実験手法によって,最近では,S. cerevisiae[5-10],Caenorhabditis elegans (C. elegans) [11,12],Drosophila melanogaster [13],ヒト[14-16]において大規模なヌクレオソーム形成DNA配列の取得がなされた.現在,ヌクレオソームポジショニングデータは確実に増えている.ヌクレオソームポジショニングは未解明ながらも,結合配列の法則性が示唆され,なおかつ結合配列のデータも増えてきており,ヌクレオソーム解析の基盤が整いつつある.
だが, DNA配列におけるヒストン結合シグナルは様々な見解があり統一されていない.ヒストンと結合する塩基配列の約半数は円状に巻きつくために物理的な制約を受けており,配列によって親和性が大きく異なる.ヒストン親和性の高いDNAには周期的にAA/AT/TAが見られるといった報告や[5],(dT,dA)nを含むS. cerevisiaeのDED1遺伝子プロモーター上にヌクレオソームが形成されるという報告がある一方[17],同じくS. cerevisiaeの転写制御領域に(dT,dA)nを導入したところ,ヌクレオソームの形成が阻害され,転写量が増加したとの報告もある[18].C. elegansにおいてはヌクレオソームを形成するDNA配列の共通する特徴はないとの報告がある一方[12],ヒトにおいてDNA配列の周期的なAT/GC含有量の変化がヌクレオソームとの親和性を高くするとの報告もある[19]. DNA配列からヌクレオソームポジショニングを予測する試みも行われており,SegalらによるAA/TTの周期性モデルによって求める手法[9],PeckhamらによるSupport Vector Machine(SVM)を用いた機械学習手法[20],Yuanらによるウェーブレット変換を用いたN-Scoreモデル[21]等があり,どれもS. cerevisiaeにおいて予測及び精度検証を行っている.SVMを用いた手法とN-Scoreモデルは精度が高い手法とされており,予測精度も同等とされている.
だが, DNA配列におけるヒストン結合シグナルは様々な見解があり統一されていない.ヒストンと結合する塩基配列の約半数は円状に巻きつくために物理的な制約を受けており,配列によって親和性が大きく異なる.ヒストン親和性の高いDNAには周期的にAA/AT/TAが見られるといった報告や[5],(dT,dA)nを含むS. cerevisiaeのDED1遺伝子プロモーター上にヌクレオソームが形成されるという報告がある一方[17],同じくS. cerevisiaeの転写制御領域に(dT,dA)nを導入したところ,ヌクレオソームの形成が阻害され,転写量が増加したとの報告もある[18].C. elegansにおいてはヌクレオソームを形成するDNA配列の共通する特徴はないとの報告がある一方[12],ヒトにおいてDNA配列の周期的なAT/GC含有量の変化がヌクレオソームとの親和性を高くするとの報告もある[19]. DNA配列からヌクレオソームポジショニングを予測する試みも行われており,SegalらによるAA/TTの周期性モデルによって求める手法[9],PeckhamらによるSupport Vector Machine(SVM)を用いた機械学習手法[20],Yuanらによるウェーブレット変換を用いたN-Scoreモデル[21]等があり,どれもS. cerevisiaeにおいて予測及び精度検証を行っている.SVMを用いた手法とN-Scoreモデルは精度が高い手法とされており,予測精度も同等とされている.
1.3 塩基組成とDNA高次構造のヌクレオソームポジショニングへの影響
DNAの高次構造がDNAとヒストンとの親和性に大きな影響を及ぼし,形成位置を左右することが報告されてきた.一般に,真確生物のDNAは負のスーパーコイル構造をとっているとされるが,この構造はヌクレオソームに内包されやすい.負のスーパーコイルの密度が高い場合,Z型DNAや三重鎖DNA,十字架構造がとられやすくなる.プロモーター領域におけるZ型DNAの形成がCSF1遺伝子の転写を促進させており,ヌクレオソームの形成阻害によって転写を活性化させているということが示唆されている[23].また,十字架構造の形成もヌクレオソームの形成を阻害するといわれている[17].
最近の報告では,2005年にヌクレオソームの配列における,規則的なAA/AT/TTの出現は,DNAの柔軟性保持するためのものだと言われている.DNAは急角度でヒストンに巻き付いているため,塩基対の水素結合がGG/GC/CCより少ないAA/AT/TTを周期的に配置することによって,柔軟性を得ているとされる.
最近の報告では,2005年にヌクレオソームの配列における,規則的なAA/AT/TTの出現は,DNAの柔軟性保持するためのものだと言われている.DNAは急角度でヒストンに巻き付いているため,塩基対の水素結合がGG/GC/CCより少ないAA/AT/TTを周期的に配置することによって,柔軟性を得ているとされる.
本研究はDNA配列からのヌクレオソームポジショニングの予測を目的とする.ヌクレオソームポジショニングの実験的な大規模取得は未だコストと時間を要する.ヌクレオソームポジショニングは部分的な取得,もしくは取得されていない生物種がほとんどである.従ってヌクレオソームポジショニングのDNA配列からの高精度予測手法の確立は,生命システムの解明に役立つ.また,ヌクレオソームポジショニングは転写因子の結合を大きく左右する要素であり,転写機構の実験デザインにも役立つだろう[22].遺伝子工学の分野においても,ヌクレオソーム形成位置は発現量を大きく左右する因子であり,発現量制御に利用されることが期待される[18].
3.1 対象
本研究ではS. cerevisiaeを対象生物種として扱った.S. cerevisiaeはヌクレオソームポジショニングの分野において,現在最も盛んに研究が行われており,最も多くの配列が得られている.
SVMに学習させるデータとしてYuanらによってChIP-chip法を用いて網羅的に得られたヌクレオソームポジショニングデータを扱う.このデータは,ヌクレオソームポジショニング予測を行う際,最も多く用いられているデータである[9][20].しかし,このデータはタイリングアレイを用いているため,配列そのものをシーケンシングによって得るChIP-seq法のデータより精度が劣っていると考えられる.そのため,本手法が正しくヌクレオソームポジショニング配列の特徴を抽出できているかを確認する精度検証に,SegalらによってChIP-chip法を用いて得られたデータを扱う.このデータも,ヌクレオソームポジショニングの研究において,使われる頻度の高いデータである[5][21].
SVMに学習させるデータとしてYuanらによってChIP-chip法を用いて網羅的に得られたヌクレオソームポジショニングデータを扱う.このデータは,ヌクレオソームポジショニング予測を行う際,最も多く用いられているデータである[9][20].しかし,このデータはタイリングアレイを用いているため,配列そのものをシーケンシングによって得るChIP-seq法のデータより精度が劣っていると考えられる.そのため,本手法が正しくヌクレオソームポジショニング配列の特徴を抽出できているかを確認する精度検証に,SegalらによってChIP-chip法を用いて得られたデータを扱う.このデータも,ヌクレオソームポジショニングの研究において,使われる頻度の高いデータである[5][21].
3.2 SVMを用いたヌクレオソームポジショニング予測手法の概要
本研究では,DNA配列からのヌクレオソームポジショニングの予測を行った.実験的に既知のヌクレオソームポジショニングデータからDNA配列の特徴を抽出し,予測を行った.特徴抽出手法として,現在最も性能の高いアルゴリズムの一つであるSVMを用いた.SVMはポジティブデータセットとネガティブデータセットを必要とする機械学習手法であり,与えられた問題(テストセット)を2クラスに分類する.ヌクレオソームポジショニング予測を,ヌクレオソームを形成するor形成しない,の2クラスへの分類問題であると考え,SVMを用いた(図1).
SVMを用いた類似の研究としてPeckhamらの手法が挙げられる[20].本手法はSVMで機械学習を行うデータセットの選別法が大きく異なる.本手法の優位性を示すため,Peckhamらと同じヌクレオソームポジショニングデータを用いた[9].
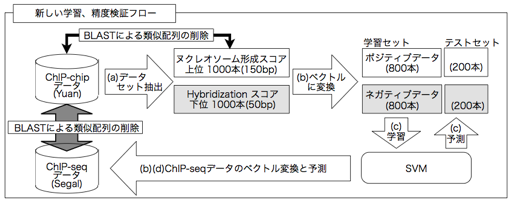
図 1 機械学習,精度検証フローの概要
SVMは学習にベクトルデータが求められる.ベクトルデータへの変換法は,データセットの配列を,配列内に含まれる長さkの配列含有率に変換する手法を用いた.本手法ではkは1〜6である(A,T,G,C,AA,AT,AG…CCCCCCそれぞれの含有率がベクトルデータとして与えられる).ベクトルの次元は41から46までを足し合わせた5460次元となった.
第一の精度検証として5-fold クロスバリデーションを用いた.機械学習時にデータセットの1/5を除外して学習させ,学習しなかったデータを正しく判別する精度を求めた.
第二の精度検証として,上記のChIP-chip法で得られたデータセットによって機械学習を行い,ChIP-seq法によって得られたデータを判別させた[5].ChIP-chip法でのデータから正しくヌクレオソームポジショニング配列の特徴を抽出できていれば,ChIP-seq法でのデータを正しく判別できるという考えのもと行った.
SVMを用いた類似の研究としてPeckhamらの手法が挙げられる[20].本手法はSVMで機械学習を行うデータセットの選別法が大きく異なる.本手法の優位性を示すため,Peckhamらと同じヌクレオソームポジショニングデータを用いた[9].
図 1 機械学習,精度検証フローの概要
研究フローの改善と自動化,ベクトル変換の高速化により大規模な予測を目指した.また,BLASTによる学習セットとテストセットの類似配列の削除により,精度検証の正当性を向上させた.
(a) ヌクレオソーム形成スコア上位1000本とHybridizationスコア下位1000本をデータセットとして抽出.
(b) SVMが扱えるベクトルデータにするため,1〜6塩基の配列含有率から成る5460次元のベクトルデータに変換.
(c) 除外しなかったデータセットにて学習し,除外したデータを判別し,クロスバリデーションとROCカーブを用いて予測
精度を検証.
(d) ChIP-chipデータによって学習させたSVMによってChIP-seqデータを判別し,クロスバリデーションにより予測精度を
検証.
ポジティブデータセットとして,ChIP-chipデータからHidden Markov Model(HMM)によるヌクレオソーム形成スコアで判定されたヌクレオソームポジショニング配列のうち[9],最もスコアの高い配列1000本を取得した.Peckhamらは単純にタイリングアレイとのHybridizationスコアの高い配列1000本を用いている.ChiP-chip法に用いられたタイリングアレイのプローブ長が50bpであるため,Peckhamらはヌクレオソームポジショニング配列本来の長さの150bpとは異なる,50bpの配列を学習させている.本手法は150bpの配列がポジティブデータセットとなっている.ネガティブデータセットとしてHybridizationスコアの最も低い1000本を取得した.タイリングアレイのプローブ長が50bpであるが,リンカー部本来の配列長は約50bpであるため問題ないと考え,これをネガティブデータセットとした.ネガティブデータセットはPeckhamらと全く同じものとなっている.(a) ヌクレオソーム形成スコア上位1000本とHybridizationスコア下位1000本をデータセットとして抽出.
(b) SVMが扱えるベクトルデータにするため,1〜6塩基の配列含有率から成る5460次元のベクトルデータに変換.
(c) 除外しなかったデータセットにて学習し,除外したデータを判別し,クロスバリデーションとROCカーブを用いて予測
精度を検証.
(d) ChIP-chipデータによって学習させたSVMによってChIP-seqデータを判別し,クロスバリデーションにより予測精度を
検証.
SVMは学習にベクトルデータが求められる.ベクトルデータへの変換法は,データセットの配列を,配列内に含まれる長さkの配列含有率に変換する手法を用いた.本手法ではkは1〜6である(A,T,G,C,AA,AT,AG…CCCCCCそれぞれの含有率がベクトルデータとして与えられる).ベクトルの次元は41から46までを足し合わせた5460次元となった.
第一の精度検証として5-fold クロスバリデーションを用いた.機械学習時にデータセットの1/5を除外して学習させ,学習しなかったデータを正しく判別する精度を求めた.
第二の精度検証として,上記のChIP-chip法で得られたデータセットによって機械学習を行い,ChIP-seq法によって得られたデータを判別させた[5].ChIP-chip法でのデータから正しくヌクレオソームポジショニング配列の特徴を抽出できていれば,ChIP-seq法でのデータを正しく判別できるという考えのもと行った.
3.3 AT/GC contentとDNA柔軟性による予測配列の検証
学習セットと本手法によって予測された配列のAT/GC contentを比較した.ヌクレオソームポジショニング予測は第一染色体似に対し,網羅的に行った.AT/GC contentの周期性はヌクレオソームポジショニングに強く影響しているため,本手法によって予測された配列も同等の周期性を持つと考え,比較を行った.各配列を中心でそろえ,AT/GC contentをウィンドウサイズ3bpにて求めた.また,離散フーリエ変換によってAT/GC contentの周期性を求めた.離散フーリエ変換にはCPANにて提供されているMath::FFTモジュールを用いた.
また,DNA柔軟性もヌクレオソームポジショニングに大きな影響を及ぼす要素と考えられているため,学習セットと予測配列のDNA柔軟性を比較した.DNAトリプレットの柔軟性はウシの膵臓DNaseIを用いて実験的に求められており[24],このデータを用いる事によってDNA柔軟性を求めた.DNA柔軟性はDNAの主溝方向への曲がりやすさの指標であり,正の値が多きほど曲がりやすく,負になるほど曲がりにくい.今回の検証ではヌクレオソームポジショニング配列を中心でそろえ,ウィンドウサイズ3bpでDNA配列上をサーチし,各トリプレットの柔軟性を求め,比較を行った.DNA柔軟性においても,離散フーリエ変換により周期性を求めた.
また,DNA柔軟性もヌクレオソームポジショニングに大きな影響を及ぼす要素と考えられているため,学習セットと予測配列のDNA柔軟性を比較した.DNAトリプレットの柔軟性はウシの膵臓DNaseIを用いて実験的に求められており[24],このデータを用いる事によってDNA柔軟性を求めた.DNA柔軟性はDNAの主溝方向への曲がりやすさの指標であり,正の値が多きほど曲がりやすく,負になるほど曲がりにくい.今回の検証ではヌクレオソームポジショニング配列を中心でそろえ,ウィンドウサイズ3bpでDNA配列上をサーチし,各トリプレットの柔軟性を求め,比較を行った.DNA柔軟性においても,離散フーリエ変換により周期性を求めた.
4.1 本手法とPeckhamの手法の精度比較
Accuracy,ROCカーブでの比較において,本手法の予測精度がPeckhamらの予測精度を上回った(図2).Accuracyでの予測精度比較では,ChIP-chipデータ,ChIP-seqデータ共にPeckhamらの手法より高い精度を出している.特にChIP-seqデータでは大きな差が出た.
ChIP-chipデータでのROCカーブによる予測精度比較では本手法のROCスコアが0.84,Peckhamらの手法が0.828と,本手法の予測精度が高い値を出した.
BLASTによる類似配列のフィルタリングにより,より正確な予測精度の値を出すことができた.フィルタリング前よりも予測精度が下がっており,不正に高い予測精度は検出されていない.学習セットとテストセットとの類似配列のフィルタリングにより,先学期とは異なる結果となっているが,本手法の予測精度がPeckhamらの予測精度より高いことに変わりは無かった.
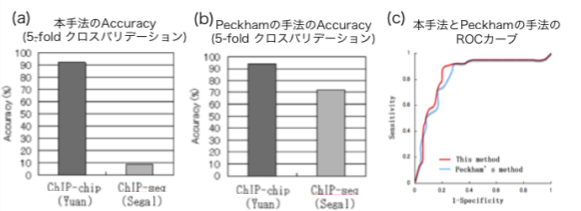
図2. 本手法とPeckhamの手法による精度比較
ChIP-chipデータでのROCカーブによる予測精度比較では本手法のROCスコアが0.84,Peckhamらの手法が0.828と,本手法の予測精度が高い値を出した.
BLASTによる類似配列のフィルタリングにより,より正確な予測精度の値を出すことができた.フィルタリング前よりも予測精度が下がっており,不正に高い予測精度は検出されていない.学習セットとテストセットとの類似配列のフィルタリングにより,先学期とは異なる結果となっているが,本手法の予測精度がPeckhamらの予測精度より高いことに変わりは無かった.
図2. 本手法とPeckhamの手法による精度比較
(a)(b) ChIP-chip法で得られるデータ及びChIP-seq法で得られたデータを,新しいフローで予測した場合のAccuracyによる精度比較.x軸がテストセットの実験手法,y軸がAccuracyである.(a)がPeckhamらの手法による予測精度,(b)が本手法による予測精度である.両方の手法共に,YuanらによるChIP-chipデータによって学習させている.本手法ではChIP-chip法が94.2%,ChIP-seq法が72.5%の精度であった.これに対し,Peckham手法では,ChIP-chip法が92.3%,ChIP-seq法が8.7%の正答率であった.
(c)ChIP-chip法によるデータを用いて,Peckhamらの手法と新しいフローによる予測精度でのROCカーブによる比較を行った.赤いラインが本手法,青いラインがPeckhamらの手法である.本手法のROCスコアは0.84,PeckhamのROCスコアは0.828であった.
(c)ChIP-chip法によるデータを用いて,Peckhamらの手法と新しいフローによる予測精度でのROCカーブによる比較を行った.赤いラインが本手法,青いラインがPeckhamらの手法である.本手法のROCスコアは0.84,PeckhamのROCスコアは0.828であった.
4.2 AT/GC contentを用いたヌクレオソームポジショニング配列の解析と予測配列の検証
ChIP-chip法により得られたヌクレオソームポジショニング配列,本手法によって予測した配列共に約5bpごとのピークが見られた(図3a,c).両方のAT/GC contentの波形は酷似している(付録 図 1).また,AT/GC contentの平均値もゲノム全体,リンカー部の配列と比べて,実験データと予測配列には同じ傾向がみられた(付録 図 2a).
離散フーリエ変換での周期性抽出では,実験データ,予測配列ともに9bp,12bp,14bpの周期でピークが見られた(図 3b,d).得られた9bpの周期は,AT/GC contentで見られた約5bpおきのピークである.AT/GC contentでは約5bpでの山,約5bpでの谷となっているため,波形の周期に変換した場合,山と谷を足し合わせた約10bpでの周期性ということになる.低い周期では実験データに2bpの周期,予測配列に1bpの周期が検出された.また,予測配列にのみ15bpの周期がみられた.実験で得られたリンカー配列ではこのような周期はみられず,第一染色体全体では実験によるリンカー配列とヌクレオソームポジショニング配列の周期を足しあわせたようなピークが見られた(付録 図 2b,c,d).
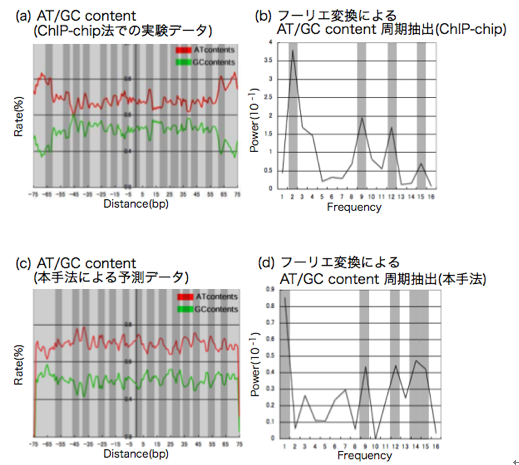
図3 学習セットと本手法によって予測された配列のAT/GC contentと離散フーリエ変換による周期性抽出
離散フーリエ変換での周期性抽出では,実験データ,予測配列ともに9bp,12bp,14bpの周期でピークが見られた(図 3b,d).得られた9bpの周期は,AT/GC contentで見られた約5bpおきのピークである.AT/GC contentでは約5bpでの山,約5bpでの谷となっているため,波形の周期に変換した場合,山と谷を足し合わせた約10bpでの周期性ということになる.低い周期では実験データに2bpの周期,予測配列に1bpの周期が検出された.また,予測配列にのみ15bpの周期がみられた.実験で得られたリンカー配列ではこのような周期はみられず,第一染色体全体では実験によるリンカー配列とヌクレオソームポジショニング配列の周期を足しあわせたようなピークが見られた(付録 図 2b,c,d).
図3 学習セットと本手法によって予測された配列のAT/GC contentと離散フーリエ変換による周期性抽出
(a)(c) ヌクレオソームポジショニング配列の中心で配列をそろえた場合のAT/GC content.x軸が含有量(%),横軸が中心からの距離である.(a)がChIP-chip法で得られたYuanらのヌクレオソームポジショニング配列のAT/GC content,(c)が本手法によって予測した配列である.両方の配列においてAT/GC contentに約5bpおきのピークが見られる(灰色が重ねられている部分).
(b)(d) AT/GC contentを離散フーリエ変換した周期の抽出.x軸が波形強度,y軸が周波数である.(b)がYuanらによって得られたChIP-chip法による配列,(d)が本手法によって予測された配列である.9bp,12bp,14bpの周期性が共通して得られている.双方ともほぼ同じ周期性がみられる(灰色で塗られている部分).
(b)(d) AT/GC contentを離散フーリエ変換した周期の抽出.x軸が波形強度,y軸が周波数である.(b)がYuanらによって得られたChIP-chip法による配列,(d)が本手法によって予測された配列である.9bp,12bp,14bpの周期性が共通して得られている.双方ともほぼ同じ周期性がみられる(灰色で塗られている部分).
4.3 DNA柔軟性によるヌクレオソームポジショニング配列の解析と予測配列の検証
ChIP-chip法による実験データ(Yuan)と予測した配列のDNA柔軟性比較では,共通するシグナルは発見できなかった(図 3).実験データにのみ27bp,28bpの部分において非常に柔軟性が低くなっている特徴がみられた.配列内のDNA柔軟性平均においても実験データと予測配列との共通性は見られ無かった.配列内における平均柔軟性は低い順に,第一染色体全体,予測配列,リンカー,学習セットとなった(付録 図3c).
離散フーリエ変換によるDNA周期性の抽出においても,共通性は見られなかった.実験データでは9bpの周期で強いピークが見られたが,予測配列においては9bpにおける周期は皆無であった.共通する周期は,6bpの部分に実験データより弱いピークが予測データにも出ている程度であった.リンカー配列では3bpの周期性が最も強く,これとも類似点は無かった (付録 図3b). 第一染色体全体のDNA柔軟性の周期は,2bp,5bp,8bpの周期が最も強く,次いで151bpの周期が見られた(付録 図 4).
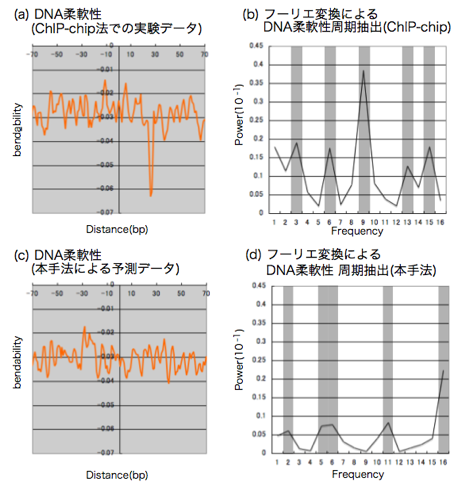
図3 学習セットと本手法によって予測された配列のDNA柔軟性と離散フーリエ変換による周期性抽出
離散フーリエ変換によるDNA周期性の抽出においても,共通性は見られなかった.実験データでは9bpの周期で強いピークが見られたが,予測配列においては9bpにおける周期は皆無であった.共通する周期は,6bpの部分に実験データより弱いピークが予測データにも出ている程度であった.リンカー配列では3bpの周期性が最も強く,これとも類似点は無かった (付録 図3b). 第一染色体全体のDNA柔軟性の周期は,2bp,5bp,8bpの周期が最も強く,次いで151bpの周期が見られた(付録 図 4).
図3 学習セットと本手法によって予測された配列のDNA柔軟性と離散フーリエ変換による周期性抽出
(a)(c)ヌクレオソーム配列を中心でそろえた場合のDNA柔軟性.(a)がChIP-chip法による実験データのDNA柔軟性,(b)が本手法により予測された配列のDNA柔軟性である.実験データは中心から27bp,28bpにDNA柔軟性が非常に低い部分が検出された.
(b)(d)学習セットと本手法によって予測配列の離散フーリエ変換を用いた周期性抽出.(b)が学習セット,(d)が予測配列である.学習セットには3bp,6bp,9bp,13bp,15bpの周期でピークが見られ,予測配列では,2bp,5bp,6bp,11bp,16bp,の周期でピークが見られた.
(b)(d)学習セットと本手法によって予測配列の離散フーリエ変換を用いた周期性抽出.(b)が学習セット,(d)が予測配列である.学習セットには3bp,6bp,9bp,13bp,15bpの周期でピークが見られ,予測配列では,2bp,5bp,6bp,11bp,16bp,の周期でピークが見られた.
5.1 議論
5-foldクロスバリデーション,ROCカーブを用いた精度予測では,本手法がPeckhamらの手法よりも,高い精度で予測できることが示された.先学期よりも予測精度の数値は低なったものの,Peckhamらの手法より本手法の予測精度が高いことに変化はなかった.また,両手法ともYuanらのChIP-chip法による同じ学習セットを用いているにも関わらず本手法の予測精度が高かった事と,異なる実験手法で得られたデータの判別精度も高い事から,本手法はPeckhamらの手法以上にヌクレオソームポジショニング配列の特徴を抽出できていると言える.
AT/GC contentによる実験データと予測配列の比較では非常に類似した結果が出た.学習させたSVMによって予測されたヌクレオソーム形成配列と学習セットのAT/GC contentが共に約9bpの周期を持っていたことから,正しく配列特徴を学習できていることが確かめられた.この周期性はS. cerevisiaeとヒトのヌクレオソーム配列において報告されており,本研究での結果を支持するものである.配列全体でのAT/GC contentの値も,ヌクレオソームポジショニング配列ではGCリッチ,リンカーではATリッチとなっており,既知の知見と一致する[19].リンカー配列からは5bpの周期が見つかり,これまでの報告にはないものである.
DNA柔軟性による比較では,実験で得られたヌクレオソームポジショニング配列と予測によって得られた配列とでは類似性が見られなかった.実験データのDNA柔軟性で最も特徴的だった,27bp,28bpの部分での柔軟性低下や,離散フーリエ変換で見られた9bpの周期性も見られなかった.本手法ではヌクレオソームポジショニング配列上のDNA柔軟性は学習されていないと考えられる.また,DNA柔軟性の配列内の平均値は,実験データによるヌクレオソームポジショニング配列が最も高く,第一染色体全体が最も低い.リンカー配列はヌクレオソームポジショニングと第一染色体全体との中間に位置しているため,ヌクレオソームポジショニングでもリンカーでも無い部分が最も曲がりにくいと考察できる.例えばスライディングのように,ヌクレオソームが移動する,ヌクレオソームポジショニングとも,リンカー部分とも判別されない部分が最も曲がりにくいということが予想される.
本研究で用いたDNA柔軟性は主溝への曲がりやすさを示すものであるため,DNAのゆがみやすさは考慮に入れられていない.DNAの柔軟性には主溝方向への角度だけでなく,平行方向へのゆがみという概念もある.ヌクレオソームポジショニングにおいては主溝方向ではなく,平行方向へのゆがみが重要になっている可能性がある.また,第一染色体のDNA柔軟性の離散フーリエ変換で見られた150bp周期はヌクレオソームによる周期である可能性もある.DNA柔軟性について深い議論を行うためには,さらなる調査が必要である.
DNA柔軟性の解析による,実験データのヌクレオソームポジショニング配列と本手法で予測したヌクレオソームポジショニング配列とで共通性は見られなかった.しかし,AT/GC contentによる共通性が見られ,また,精度検証による予測精度の高さから,現実的な精度で予測できるだけの特徴は抽出できたと著者は考えている.
AT/GC contentによる実験データと予測配列の比較では非常に類似した結果が出た.学習させたSVMによって予測されたヌクレオソーム形成配列と学習セットのAT/GC contentが共に約9bpの周期を持っていたことから,正しく配列特徴を学習できていることが確かめられた.この周期性はS. cerevisiaeとヒトのヌクレオソーム配列において報告されており,本研究での結果を支持するものである.配列全体でのAT/GC contentの値も,ヌクレオソームポジショニング配列ではGCリッチ,リンカーではATリッチとなっており,既知の知見と一致する[19].リンカー配列からは5bpの周期が見つかり,これまでの報告にはないものである.
DNA柔軟性による比較では,実験で得られたヌクレオソームポジショニング配列と予測によって得られた配列とでは類似性が見られなかった.実験データのDNA柔軟性で最も特徴的だった,27bp,28bpの部分での柔軟性低下や,離散フーリエ変換で見られた9bpの周期性も見られなかった.本手法ではヌクレオソームポジショニング配列上のDNA柔軟性は学習されていないと考えられる.また,DNA柔軟性の配列内の平均値は,実験データによるヌクレオソームポジショニング配列が最も高く,第一染色体全体が最も低い.リンカー配列はヌクレオソームポジショニングと第一染色体全体との中間に位置しているため,ヌクレオソームポジショニングでもリンカーでも無い部分が最も曲がりにくいと考察できる.例えばスライディングのように,ヌクレオソームが移動する,ヌクレオソームポジショニングとも,リンカー部分とも判別されない部分が最も曲がりにくいということが予想される.
本研究で用いたDNA柔軟性は主溝への曲がりやすさを示すものであるため,DNAのゆがみやすさは考慮に入れられていない.DNAの柔軟性には主溝方向への角度だけでなく,平行方向へのゆがみという概念もある.ヌクレオソームポジショニングにおいては主溝方向ではなく,平行方向へのゆがみが重要になっている可能性がある.また,第一染色体のDNA柔軟性の離散フーリエ変換で見られた150bp周期はヌクレオソームによる周期である可能性もある.DNA柔軟性について深い議論を行うためには,さらなる調査が必要である.
DNA柔軟性の解析による,実験データのヌクレオソームポジショニング配列と本手法で予測したヌクレオソームポジショニング配列とで共通性は見られなかった.しかし,AT/GC contentによる共通性が見られ,また,精度検証による予測精度の高さから,現実的な精度で予測できるだけの特徴は抽出できたと著者は考えている.
5.2 展望
本研究によってS. cerevisiaeにおける,予測手法を確立できたといえる.そこで興味の対象となるのは,他の生物種における予測である.これには2つのケースがあり,対象となる生物種の一部のヌクレオソームポジショニング配列を学習させて予測するケースと,そもそも全く異なる生物種のヌクレオソームを学習させて,対象生物種のヌクレオソームポジショニングを予測するケースである.前者は部分的にしかヌクレオソームポジショニング配列がとられていない生物種での予測を可能とし,後者はまったくヌクレオソームポジショニング配列がえられていない生物種での予測を可能とする.特に後者は,真核生物の持つ種を超えたヌクレオソームポジショニングのルールの存在を示唆することが可能である.
また,S. cerevisiaeにおける精度も向上する余地がある.今回の手法では1〜6bpの配列含有率であったが,1〜12bpの配列含有率といったように,学習セットにおけるベクトルの次元数を高める事で,予測精度をさらに向上させる事ができるだろう.DNA柔軟性については,平行方向へのゆがみというパラメータを用いることによって,ヌクレオソームポジショニング配列における特徴を得られる可能性がある.
また,S. cerevisiaeにおける精度も向上する余地がある.今回の手法では1〜6bpの配列含有率であったが,1〜12bpの配列含有率といったように,学習セットにおけるベクトルの次元数を高める事で,予測精度をさらに向上させる事ができるだろう.DNA柔軟性については,平行方向へのゆがみというパラメータを用いることによって,ヌクレオソームポジショニング配列における特徴を得られる可能性がある.
本研究は,僕の周りにいる多くの方々のご好意で行うことが可能となりました.友人や先輩,教員のみなさんのご指導に支えられています.多大な迷惑をかけてしまいつつも,暖かくご指導しつづけてくださった首長の斎藤輪太郎さんや,アドバイザーの北川統之さんなどさまざまな方の助力をいただきました.また,冨田教授にはこのような研究の場を与えていただきました.そして、森泰吉郎記念研究振興基金「研究育成費」の補助により、快適な研究環境をそろえる事ができました。この場を借りて,感謝の意を表させていただきます.本当にありがとうございました.
[1] Luger, K., et al. (1997) Crystal structure of the nucleosome core particle at 2.8 Å resolution, Nature
389, 251-260.
[2] Drew, H.R. and Travers, A.A. (1985) DNA bending and its relation to nucleosome positioning, J Mol
Biol. 186, 773–790.
[3] Lee, C.K., et al. (2004) Evidence for nucleosome depletion at active regulatory regions
genome-wide, Nat Genet, 36, 900-905.
[4] Kornberg, R.D. (1974) Chromatin Structure: A Repeating Unit of Histones and DNA, Science, 184,
868-871.
[5] Segal, E., et al. (2006) A genomic code for nucleosome positioning, Nature, 442, 772-778.
[6] Bernstein, B.E., et al. (2004) Global nucleosome occupancy in yeast, Genome Biol, 5, R62.
[7] Lee, C.K., et al. (2004) Evidence for nucleosome depletion at active regulatory regions
genome-wide, Nat Genet, 36, 900-905.
[8] Pokholok, D.K., et al. (2005) Genome-wide map of nucleosome acetylation and methylation in yeast,
Cell, 122, 517-527.
[9] Yuan, G.C., et al. (2005) Genome-scale identification of nucleosome positions in S. cerevisiae,
Science, 309, 626-630.
[10] Albert, I., et al. (2007) Translational and rotational settings of H2A.Z nucleosomes across the
Saccharomyces cerevisiae genome, Nature, 446, 572-576.
[11] Johnson, S.M., et al. (2006) Flexibility and constraint in the nucleosome core landscape of
Caenorhabditis elegans chromatin. Genome Res, 16, 1505-1516.
[12] Valouea, A., et al, (2008) A high-resolution, nucleosome position map of C. elegans reveals a lack
of universal sequence-dictated positioning, Genome Res, 18, 1051-1063.
[13] Mito, Y., et al. (2005) Genome-scale profiling of histone H3.3 replacement patterns, Nat Genet,
37, 1090-1097.
[14] Ozsolak, F., et al. (2007) High-throughput mapping of the chromatin structure of human
promoters, Nat Biotechnol, 25, 244-248.
[15] Heintzman, N.D., et al. (2007) Distinct and predictive chromatin signatures of transcriptional
promoters and enhancers in the human genome, Nat Genet, 39, 311-318.
[16] Barski, A., et al. (2007) Highresolution profiling of histone methylations in the human genome.
Cell, 129, 823–837.
[17] Ohyama, T. (2005) The role of unusual DNA structures in chromatin organization for
transcription, in DNA Conformation and Transcription, pp. 177-188.
[18] Morohashi, N., et al. (2006) Effect of sequence-directed nucleosome disruption on cell-type-
specific repression by alpha2/Mcm1 in the yeast genome, Eukaryot Cell, 5, 1925-33.
[19] Chung, H.R. and Vingron, M. (2008) Sequence-dependent Nucleosome Positioning, J Mol Biol,
doi:10.1016/j.jmb.2008.11.049.
[20] Peckham, E.P., et al. (2007) Nucleosome positioning signals in genomic DNA, Genome Res, 17,
1170-1177.
[21] Yuan, G.C. and Liu, J.S. (2008) Genomic sequence is highly predictive of local nucleosome
depletion, PLoS Comput Biol, 4, 0164-0174.
[22] Narlikar, L., et al. (2007) Nucleosome occupancy information improves de novo motif discovery.
RECOMB, 107-121.
[23] Liu, R., et al. (2001) Regulation of CSF1 promoter by the SWI/SNF-like BAF complex, Cell, 106,
309-318.
[24] Brukner, I. et al, (1995) Sequence-dependent bending propensity of DNA as revealed by DNase 1:
parameters for trinucleotides, EMBO, 14, 1812-1818.
389, 251-260.
[2] Drew, H.R. and Travers, A.A. (1985) DNA bending and its relation to nucleosome positioning, J Mol
Biol. 186, 773–790.
[3] Lee, C.K., et al. (2004) Evidence for nucleosome depletion at active regulatory regions
genome-wide, Nat Genet, 36, 900-905.
[4] Kornberg, R.D. (1974) Chromatin Structure: A Repeating Unit of Histones and DNA, Science, 184,
868-871.
[5] Segal, E., et al. (2006) A genomic code for nucleosome positioning, Nature, 442, 772-778.
[6] Bernstein, B.E., et al. (2004) Global nucleosome occupancy in yeast, Genome Biol, 5, R62.
[7] Lee, C.K., et al. (2004) Evidence for nucleosome depletion at active regulatory regions
genome-wide, Nat Genet, 36, 900-905.
[8] Pokholok, D.K., et al. (2005) Genome-wide map of nucleosome acetylation and methylation in yeast,
Cell, 122, 517-527.
[9] Yuan, G.C., et al. (2005) Genome-scale identification of nucleosome positions in S. cerevisiae,
Science, 309, 626-630.
[10] Albert, I., et al. (2007) Translational and rotational settings of H2A.Z nucleosomes across the
Saccharomyces cerevisiae genome, Nature, 446, 572-576.
[11] Johnson, S.M., et al. (2006) Flexibility and constraint in the nucleosome core landscape of
Caenorhabditis elegans chromatin. Genome Res, 16, 1505-1516.
[12] Valouea, A., et al, (2008) A high-resolution, nucleosome position map of C. elegans reveals a lack
of universal sequence-dictated positioning, Genome Res, 18, 1051-1063.
[13] Mito, Y., et al. (2005) Genome-scale profiling of histone H3.3 replacement patterns, Nat Genet,
37, 1090-1097.
[14] Ozsolak, F., et al. (2007) High-throughput mapping of the chromatin structure of human
promoters, Nat Biotechnol, 25, 244-248.
[15] Heintzman, N.D., et al. (2007) Distinct and predictive chromatin signatures of transcriptional
promoters and enhancers in the human genome, Nat Genet, 39, 311-318.
[16] Barski, A., et al. (2007) Highresolution profiling of histone methylations in the human genome.
Cell, 129, 823–837.
[17] Ohyama, T. (2005) The role of unusual DNA structures in chromatin organization for
transcription, in DNA Conformation and Transcription, pp. 177-188.
[18] Morohashi, N., et al. (2006) Effect of sequence-directed nucleosome disruption on cell-type-
specific repression by alpha2/Mcm1 in the yeast genome, Eukaryot Cell, 5, 1925-33.
[19] Chung, H.R. and Vingron, M. (2008) Sequence-dependent Nucleosome Positioning, J Mol Biol,
doi:10.1016/j.jmb.2008.11.049.
[20] Peckham, E.P., et al. (2007) Nucleosome positioning signals in genomic DNA, Genome Res, 17,
1170-1177.
[21] Yuan, G.C. and Liu, J.S. (2008) Genomic sequence is highly predictive of local nucleosome
depletion, PLoS Comput Biol, 4, 0164-0174.
[22] Narlikar, L., et al. (2007) Nucleosome occupancy information improves de novo motif discovery.
RECOMB, 107-121.
[23] Liu, R., et al. (2001) Regulation of CSF1 promoter by the SWI/SNF-like BAF complex, Cell, 106,
309-318.
[24] Brukner, I. et al, (1995) Sequence-dependent bending propensity of DNA as revealed by DNase 1:
parameters for trinucleotides, EMBO, 14, 1812-1818.
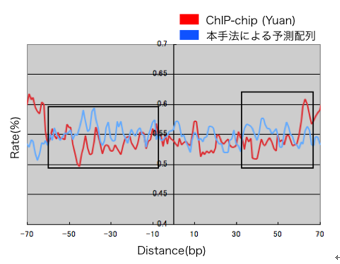
付録 図1 ChIP-chipによって得られたヌクレオソームポジショニング配列と本手法によって予測されたヌクレオソームポジショニング配列のAT contentによる比較
x軸が中心を0として各ヌクレオソームポジショニング配列をそろえた場合の距離(bp),y軸が含有量(%)である.類似した波形を描いており,特に黒い四角で囲われた部分のパターンが酷似している.
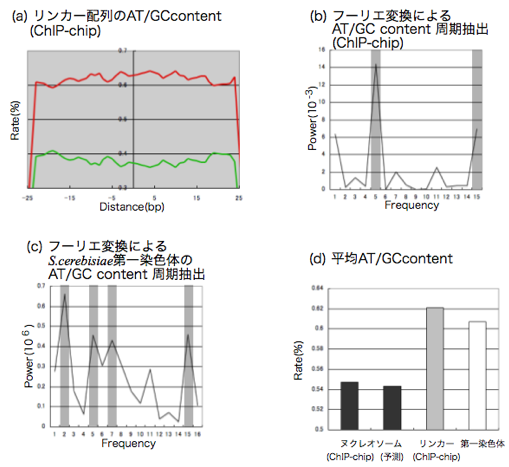
付録 図2 ChIP-chip法で得られたリンカー配列と,第一染色体全体におけるAT/GC contentの比較
(a) ChIP-chip法で得られたリンカー配列のAT/GC content.x軸は中心を0として各ヌクレオソームポジショニング配列をそろえた場合の距離(bp),y軸は含有量(%)である.
(b) ChIP-chip法で得られたリンカー配列のAT/GC contentから離散フーリエ変換によって周期性を抽出したもの.5bpと15bpの周期にピークが見られる(灰色で塗られた部分).
(c) S. cerevisiaeにおける第一染色体AT/GC contentから離散フーリエ変換によって周期性を抽出したもの.2bp,5bp,7bp,15bpの周期にピークが見られる(灰色で塗られた部分).
(d) ヌクレオソーム,リンカー,第一染色体における配列内のAT content平均の比較.リンカー部分が最もATリッチであり,ヌクレオソームはChIP-chip法による実験データと本手法による予測された配列データは共にAT contentが低い.
(b) ChIP-chip法で得られたリンカー配列のAT/GC contentから離散フーリエ変換によって周期性を抽出したもの.5bpと15bpの周期にピークが見られる(灰色で塗られた部分).
(c) S. cerevisiaeにおける第一染色体AT/GC contentから離散フーリエ変換によって周期性を抽出したもの.2bp,5bp,7bp,15bpの周期にピークが見られる(灰色で塗られた部分).
(d) ヌクレオソーム,リンカー,第一染色体における配列内のAT content平均の比較.リンカー部分が最もATリッチであり,ヌクレオソームはChIP-chip法による実験データと本手法による予測された配列データは共にAT contentが低い.
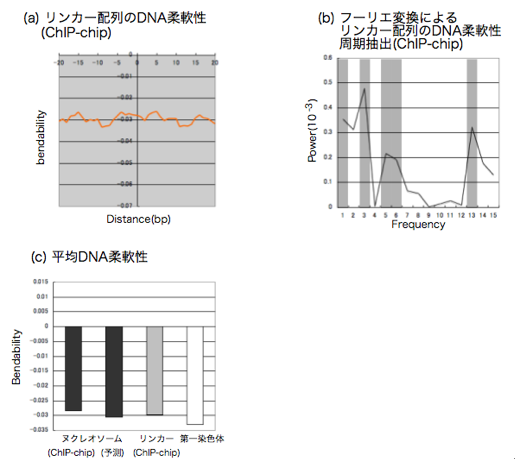
付録 図3 ChIP-chip法で得られたリンカー配列と,第一染色体全体におけるDNA柔軟性の比較
(a) ChIP-chip法で得られたリンカー配列のDNA柔軟性.x軸は配列の中心を0として各ヌクレオソームポジショニング配列をそろえた場合の距離(bp),y軸は柔軟性である.
(b) ChIP-chip法で得られたリンカー配列のDNA柔軟性から離散フーリエ変換によって周期性を抽出したもの.X軸が周期,y軸が強度である.1bp, 3bp,5bp,6bp,13bpの周期にピークが見られる(灰色で塗られた部分).
(c) ヌクレオソーム,リンカー,第一染色体における配列内のDNA柔軟性平均の比較.x軸が配列の種類,y軸が柔軟性である.第一染色体の平均が最も主溝方向への柔軟性が低い.ChIP-chip法から得られたヌクレオソームポジショニング配列はリンカーより柔軟性が高い.予測配列は第一染色体の次に柔軟性が低くなっている.
(b) ChIP-chip法で得られたリンカー配列のDNA柔軟性から離散フーリエ変換によって周期性を抽出したもの.X軸が周期,y軸が強度である.1bp, 3bp,5bp,6bp,13bpの周期にピークが見られる(灰色で塗られた部分).
(c) ヌクレオソーム,リンカー,第一染色体における配列内のDNA柔軟性平均の比較.x軸が配列の種類,y軸が柔軟性である.第一染色体の平均が最も主溝方向への柔軟性が低い.ChIP-chip法から得られたヌクレオソームポジショニング配列はリンカーより柔軟性が高い.予測配列は第一染色体の次に柔軟性が低くなっている.
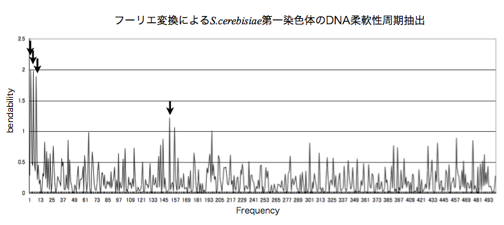
付録 図4 S. cerevisiaeにおける第一染色体DNA柔軟性からの離散フーリエ変換による周期性抽出
x軸が周期(bp),y軸が柔軟性である.2bp,5bp,8bpの周期が強くみられ,次いで151bpの周期にピークが見られる(黒い矢印)<S. cerevisiaebr>