Histone modifications play an important role in gene regulation. Acetylation of histone 3 lysine 9 (H3K9ac) is generally associated with transcription initiation and unfolded chromatin, thereby positively influencing gene expression. Deep sequencing of the 5-prime ends of gene transcripts using deepCAGE delivers detailed information about the architecture and expression level of gene promoters. The combination of H3K9ac ChIP-chip and deepCAGE in a myeloid leukemia cell line (THP-1) allowed us to study the spatial distribution of H3K9ac around promoters using a novel clustering approach. The promoter classes were analyzed for association with relevant genomic sequence features.
Figure 1 (A) shows H3K9 acetylation and gene transcription start sites in ENCODE region ENr333 as an example of how H3K9ac is concentrated around deepCAGE promoters and gene starts. Indeed, H3K9ac is localized around transcription start sites throughout the entire human genome [1][2]. A genome-wide histogram of H3K9ac around deepCAGE TSS, shown in fig. 1 (B), illustrates this on a genome-wide scale. H3K9ac has a characteristic bimodal distribution around the TSSs, with one single peak upstream of the TSS, a stronger single peak downstream of the TSS, and depletion right on the TSS. H3K9ac level right on the TSS is low because core promoters are depleted in nucleosomes [2][3]. This bimodal distribution has been described in several previous studies [4][5][6].
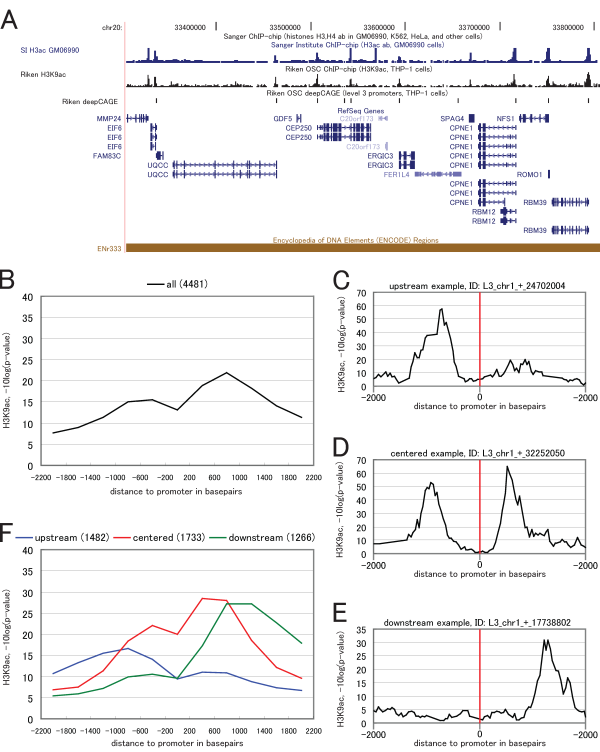
However, when inspecting the H3K9ac levels around individual promoters, the distribution of acetylation level often does not resemble the average genome-wide situation: around individual promoters the H3K9ac level may be more concentrated upstream (fig. 1 (C)) or downstream (fig. 1 (E)) of the promoter, may show a distribution which resembles the genome-wide distribution (fig. 1 (D)), or have other configurations.
We performed a clustering of 4,481 promoters according to their surrounding H3K9ac signal and analyzed the clustered promoters for association with different sequence features. The clustering revealed three groups with major H3K9ac signal upstream, centered and downstream of the promoter. Narrow single peak promoters tend to have a concentrated activity of H3K9ac in the upstream region, while broad promoters tend to have a concentrated activity of H3K9ac and RNA polymerase II binding in the centered and downstream regions. A subset of promoters with high gene expression level, compared to subsets with low and medium gene expression, shows dramatic increase in H3K9ac activity in the upstream cluster only; this may indicate that promoters in the centered and downstream clusters are predominantly regulated at post-initiation steps. Furthermore, the upstream cluster is depleted in CpG islands and more likely to regulate un-annotated genes.
The analysis of the upstream, centered and downstream clusters showed a significant bias towards promoters with different characteristics: the upstream cluster is biased towards putative novel promoters and single peak promoters. We propose that it may be regulated primarily during the initiation phase of transcription. The downstream cluster, on the other hand, is enriched in known genes, CpG islands, and broad promoters. Here we propose that regulation of promoters in the centered and downstream clusters occurs mainly in the post-initiation phase of transcription. Repeat elements are more likely to occur on core promoters with increased gene expression level, but there is no bias of repeat elements to any particular cluster. The main findings of our study are valid using experimental data from THP-1 cells in two different stages of differentiation, meaning that the number of genes changing their acetylation state during the 96 hours of differentiation is small.
Our findings suggest a functional link between the spatial distribution of H3K9 acetylation and genomic as well as transcriptomic features. Promoters belonging to the centered and downstream clusters appear similar in characteristics and are associated to features previously identified as hallmarks for ubiquitously expressed housekeeping genes (CpG islands, broad promoter shape), and accordingly are more likely to correspond to previously identified protein coding genes. In contrast, the upstream cluster is enriched in peak promoters when compared to the other two clusters, and depleted in genes overlapping with CpG islands; these features are commonly seen in promoters of genes specific to distinct tissues and cell types. The well defined TSSs of peak promoters, and distinct conditions under which they are expressed, are indicative of strict mechanisms for their regulation, and spatial distribution of open chromatin may constitute an additional mode of regulation of these genes. Conversely, an open chromatin configuration downstream of the core promoter (as observed in the centered and downstream clusters) may be either favourable for, or a consequence of, transcription from less well defined TSSs (i.e. broad promoters). The precise mechanisms of this suggested additional mode of regulation remain to be elucidated.
The results of this study have been submitted to BMC Genomics, the manuscript is now editorially accepted [7].
[1] Liang G, Lin JC, Wei V, Yoo C, Cheng JC, Nguyen CT, Weisenberger DJ, Egger G, Takai D, Gonzales FA, Jones PA: Distinct localization of histone H3 acetylation and H3-K4 methylation to the transcription start sites in the human genome. Proc Natl Acad Sci U S A 2004, 101:7357-7362
[2] Nishida H, Suzuki T, Kondo S, Miura H, Fujimura Y, Hayashizaki Y: Histone H3 acetylated at lysine 9 in promoter is associated with low nucleosome density in the vicinity of transcription start site in human cell. Chromosome Res 2006, 14:203-211
[3] Smith AE, Chronis C, Christodoulakis M, Orr SJ, Lea NC, Twine NA, Bhinge A, Mufti GJ, Thomas: Epigenetics of human T cells during the G0-G1 transition. Genome Res 2009, 19:1325-1337
[4] Roh T, Cuddapah S, Zhao K: Active chromatin domains are defined by acetylation islands revealed by genome-wide mapping. Genes Dev 2005, 19:542-552
[5] Koch CM, Andrews RM, Flicek P, Dillon SC, Karaoz U, Clelland GK, Wilcox S, Beare DM, Fowler JC, Couttet P, James KD, Lefebvre GC, Bruce AW, Dovey OM, Ellis PD, Dhami P, Langford CF, Weng Z, Birney E, Carter NP, Vetrie D, Dunham I: The landscape of histone modifications across 1% of the human genome in five human cell lines. Genome Res 2007, 17:691-707
[6] Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh T, Peng W, Zhang MQ, Zhao K: Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet 2008, 40:897-903
[7] Anton Kratz, Erik Arner, Rintaro Saito, Atsutaka Kubosaki, Jun Kawai, Harukazu Suzuki, Piero Carninci, Takahiro Arakawa, Masaru Tomita, Yoshihide Hayashizaki and Carsten O Daub: Core promoter structure and genomic context reflect histone 3 lysine 9 acetylation patterns. BMC Genomics 2010, Editorially Accepted (4/7/2010)