最小ゲノムにおける最適な遺伝子の配置方法に関する研究
政策・メディア研究科 修士課程1年 池上慶太
要旨
生物種によってゲノムサイズや遺伝子数は異なり,生存環境によっても生体機能を維持するために必要な遺伝子数は変化する.大腸菌は約400万塩基数、約4000個の遺伝子から構成されており,必ずしもすべての遺伝子が実験室環境下で必須ではないことが非必須遺伝子として同定されている.ある条件下で必須でないことを突き詰めると,生物を構成する最小の遺伝子セットを定義することができる.ここでは,このゲノムを最小ゲノムという.最小ゲノムを構成する遺伝子についてこれまでに多くの予測研究がなされているものの,実験的な実証はなされていない.そのひとつの原因として転写因子が考慮されていないことが挙げられる.本研究では,大腸菌における大規模代謝ネットワークにおける転写因子の取りうる可能性について探索した.代謝モデルはiJR904を使用し,代謝流束解析から発現制御の取りうるパターンを定量的に評価するためにバイオマスを予測した.まず,反応速度論にもとづき酵素濃度から反応速度を計算する方程式を導いた.次に,マイクロアレイデータをタンパク質存在量へ変換し,変換したタンパク質濃度を方程式へ代入し代謝反応における各反応速度を計算した.各反応速度によって代謝流束解析における解空間を再定義することで,バイオマスを予測した.ただし,このとき,発現量の大きさに応じてiJR904に含まれる遺伝子をグループ化し,グループ内に含まれる遺伝子の発現量を均一化した.結果として,グループ数を大きくし,転写単位を大きくするとバイオマスが減少する様子を予測できた.そして,バイオマスを一定量だけ保証したときの転写因子数と大腸菌における転写因子数に一定の整合性を見出すことができた.将来的に,転写因子を考慮した最小ゲノムのゲノムデザインが可能になることが期待される.ただし,バイオマスの予測結果が妥当であるか今後さらに検証する予定である.
キーワード:代謝流束解析,発現制御,物質生産能の定量的評価,タンパク質絶対量,反応速度論
1. はじめに
細胞を合成することは生物学における大きな挑戦のひとつである.細胞を構成する要素のひとつで遺伝情報をになっているゲノムの人工合成は近年技術が構築されてきている (Itaya, et al., 2008; Gibson, et al., 2010).ゲノムにはさまざまなタンパク質,非タンパク質をコードする遺伝子が存在するが,一定の生育条件下ではすべての遺伝子が必要とされていない (Koonin, 2003).ここで,「では一体いくつの遺伝子があれば生物は生物たりうるのか」という問いが生じる.このような条件を兼ね備えたゲノムを最小ゲノムといい,多くの研究者を魅了している.最小ゲノムを構成する遺伝子の候補は,複数の生物種で保存されている遺伝子の探索,必須遺伝子の探索,自然界においてゲノムサイズが小さい微生物のゲノムアノテーションなどを参考にして提唱されており,最小ゲノムを構成する遺伝子数は約150~350個が妥当な範囲であると考えられている (Moya, et al., 2009).ここで,遺伝子の転写機構について考えると,大腸菌では175個の転写因子が細胞全体の発現制御に関わっているとされる (Gama-Castro, et al., 2011).最小ゲノムを構成する遺伝子は主に代謝関連遺伝子のほかに,複製,修復,転写,翻訳に関わるものが候補として挙げられている (Gil, et al., 2004) が,これら遺伝子が十分に発現制御されるための転写因子は考慮されていない.最小ゲノムを機能による集合体ととらえると,複数の遺伝子回路を組み合わせ,集積することによって構築できると仮定できる.遺伝子回路とは遺伝子工学の技術を用いて目的の機能を果たす小規模な遺伝子群のことである.PurnickとWeissの報告によると,2000年から2008年にかけて合成生物学の発展とともに多くの遺伝子回路がつくらたものの,遺伝子回路に含まれるプロモーターの数はそれほど増加しておらず,遺伝子回路の複雑さは頭打ちになっていると指摘する (Purnick and Weiss, 2009).その他に,遺伝子回路の合成においてプロモーターとの関わりとして,変異の蓄積によるプロモーター機能の失活 (Elowitz and Leibler, 2000),宿主によるプロモーター強度の違いと遺伝子におよぼすプロモーター強度のバランス加減といった難しさが報告されている (Ellis, et al., 2009).このように,小規模な遺伝子群の遺伝子回路にあっても発現制御が困難である現状を考慮すると,最小ゲノムにおいて転写因子が含まれていないということは,宿主細胞内で機能する最小ゲノムの構築もまた困難なのではないかと考える.
そこで,本研究では大腸菌における大規模代謝モデルを用いて,発現制御の取りうるパターンについて明らかにする.生物学的な要素として,代謝経路,シグナル伝達,発現調節は時間的な要因,空間的な要因を受けにくいため,しばし静的なネットワークとして表現される (Reed, et al., 2006).特に,代謝ネットワークは,Arakawaらによってゲノム配列から高確率で再現されており,化学量論行列がバランス方程式にもとづいて構築されている (Arakawa, et al., 2006).代謝反応は幾つもの素反応が連続的に絡み合った反応系であるため,すべての素反応に対するバランス方程式を行列としてまとめたものが,化学量論行列となる (Edwards and Palsson, 1998).このことから,グルーバルな代謝経路が明らかであれば,各素反応のバランス方程式から大規模代謝ネットワークを大規模な化学量論行列として表現することができる.これを応用して,Palssonらは細胞内における動的な反応パラメターを使用することなく,線形代数的に代謝反応を解析する手法として代謝流束解析を開発した (Reed and Palsson, 2003).代謝流束解析では目的関数にバイオマスや生成物といった任意の最適化する反応速度を設定可能であるが,in silico による予測と実験的に測定された収量は妥当な類似度を大腸菌を用いた研究で報告されている (Edwards and Palsson, 2000; Fong and Palsson, 2004).よって,代謝流束解析は,動的なパラメターを用いない線形的な手法であるものの,細胞内の増殖を定量的に評価することに関して有効な手法であることがうかがえる.本研究における発現制御の取りうるパターンの探索においても,代謝流束解析を用いることで,各取りうるパターンを採用したときの細胞の増殖をバイオマスとして定量的に予測できる.以上より,本研究の目的と新規性をまとめる.研究の目的は,大腸菌における大規模代謝モデルを用いて,発現制御の取りうるパターンと実現可能性について解析することを目的とする.研究の新規性は,転写因子が考慮されていない最小ゲノムを構成する遺伝子候補から,いくつの転写因子を考慮すると一定のバイオマスを保証することが出来るのか示すことである.
代謝流束解析の新規拘束条件を設定するために,細胞内のmRNA量とタンパク質量は比例するという仮定のもと,まず遺伝子発現量を細胞内タンパク質量に変換した.ただし,細胞質中のタンパク質の定量的な測定データがあるタンパク質は変換から除外した.次に反応速度論にもとづき,置換したタンパク質量を用いて代謝経路の各素反応における反応速度を導いた.ここで導出した反応速度を各反応における反応速度の上限値として代謝流束解析において再設定した.最後に,マイクロアレイデータを降べき順に並び替え,順番に遺伝子を一定数ごとグループ化し,グループ内の遺伝子の発現量を平均値で均一化した.この発現量を均一化したグループから代謝流束解析と線形計画法によってバイオマスを予測した.
2. 対象と手法
まず「2.1. マイクロアレイデータから反応速度の数学的な導出」にて,発現量データから反応速度が予測可能であることを数学的に導いた.次に「2.2. マイクロアレイデータとタンパク質存在量との相関関係」において,反応速度論にもとづく数学的証明の前提条件となる細胞内mRNA量,すなわち発現量とタンパク質存在量に相関があるのかを解析した.そして「2.3. マイクロアレイデータのグループ化と収量の予測」において,発現量の最適化方法と反応速度の制約条件を再定義し,目的関数としてバイオマスを予測した.
2.1. マイクロアレイデータから反応速度の数学的な導出
まず,質量作用則より,反応速度Vは
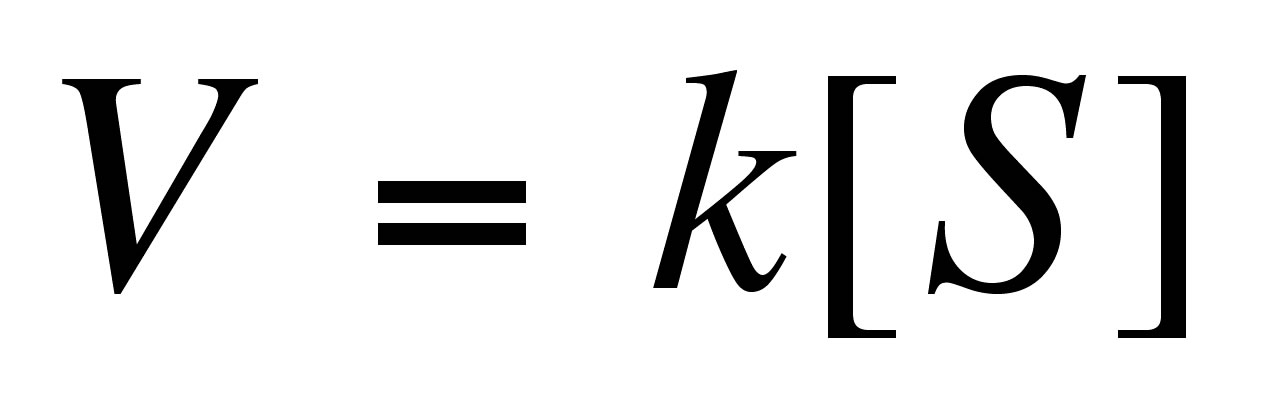 式 (1)
式 (1)
として表される.このとき,反応速度Vと反応速度定数kについての比例関係が導かれる.
 式 (2)
式 (2)
ミカエリスメンテン式より,反応速度Vは
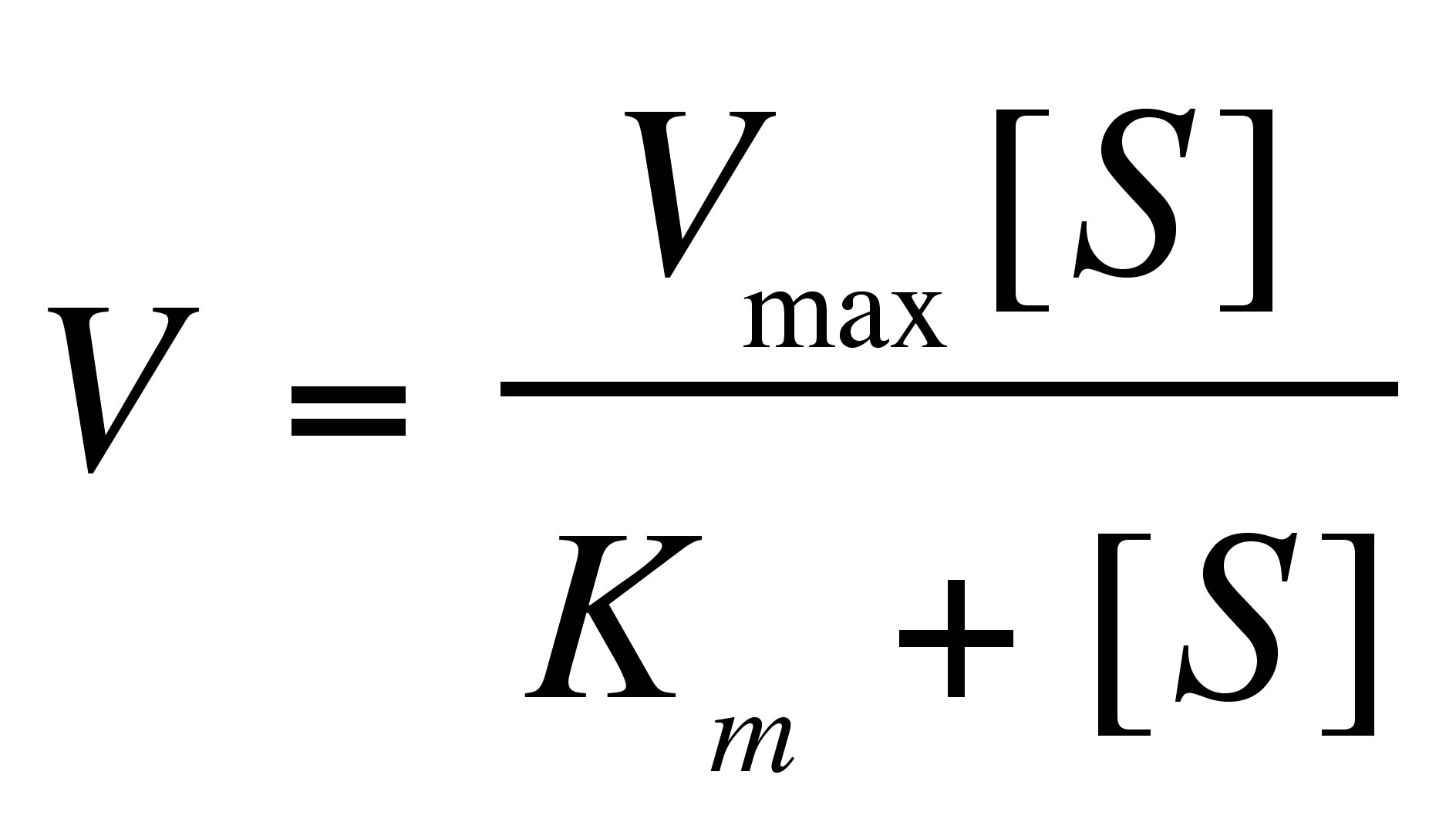 式 (3)
式 (3)
として表され、最大反応速度Vmaxは
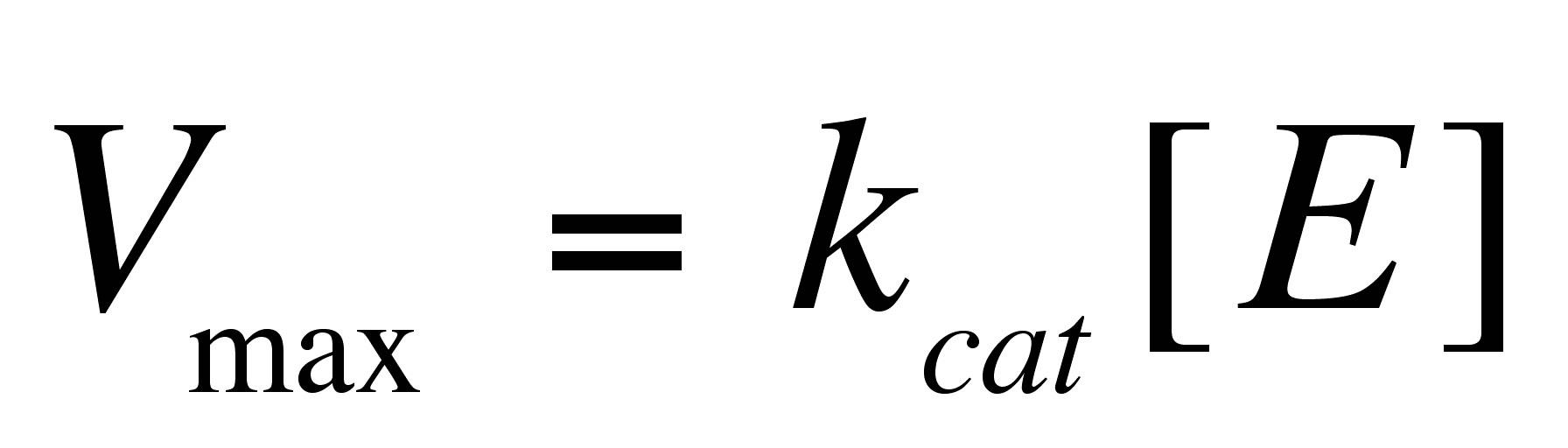 式 (4)
式 (4)
である.ここから,反応速度Vと酵素濃度[E]についての比例関係が導かれる.
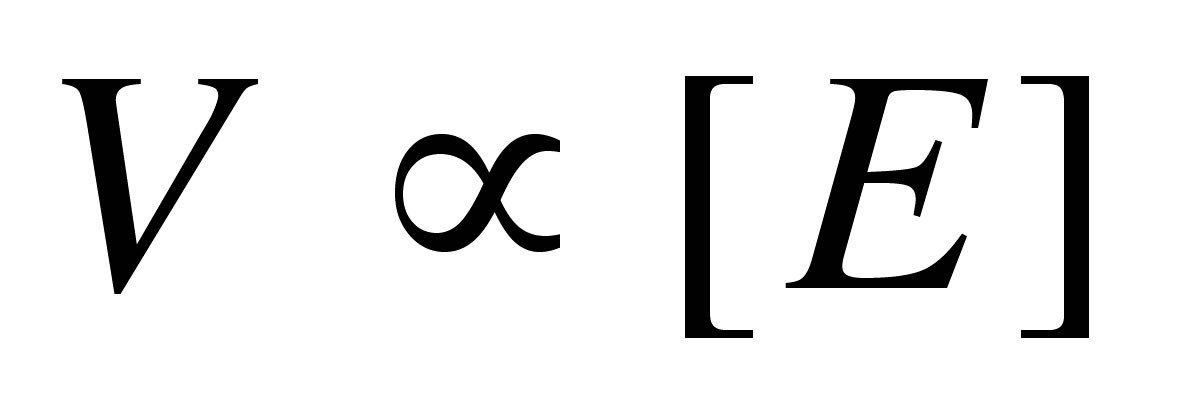 式 (5)
式 (5)
式 (2) と式 (5) より,反応速度定数kと酵素濃度[E]について以下の関係式が成り立つ.
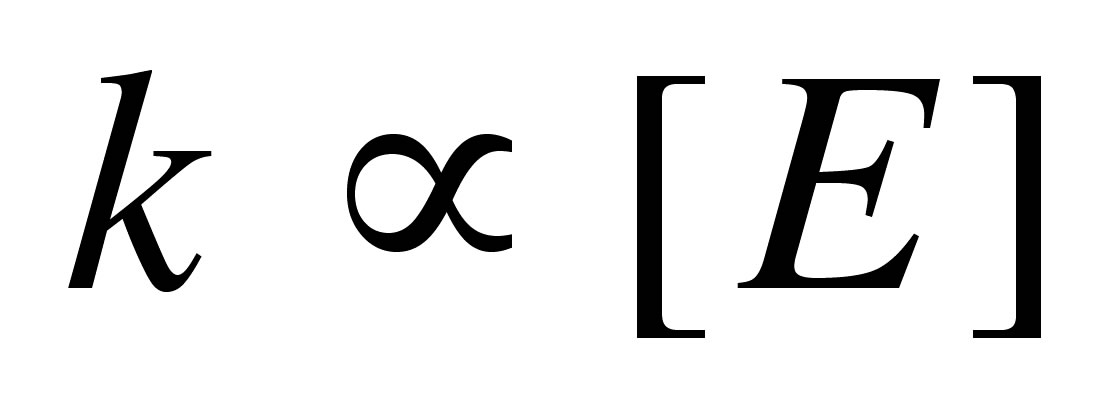 式 (6)
式 (6)
質量作用則より導かれた反応速度VをVma,ミカエリスメンテン式より導かれた反応速度VをVmmとおき,両者の比を求めると
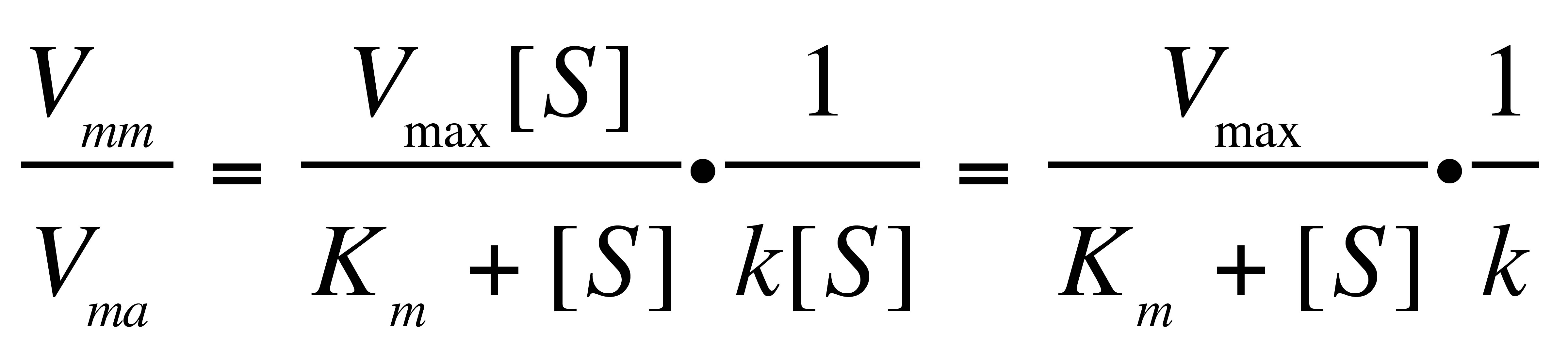 式 (7)
式 (7)
反応速度定数kについて解くことができる.
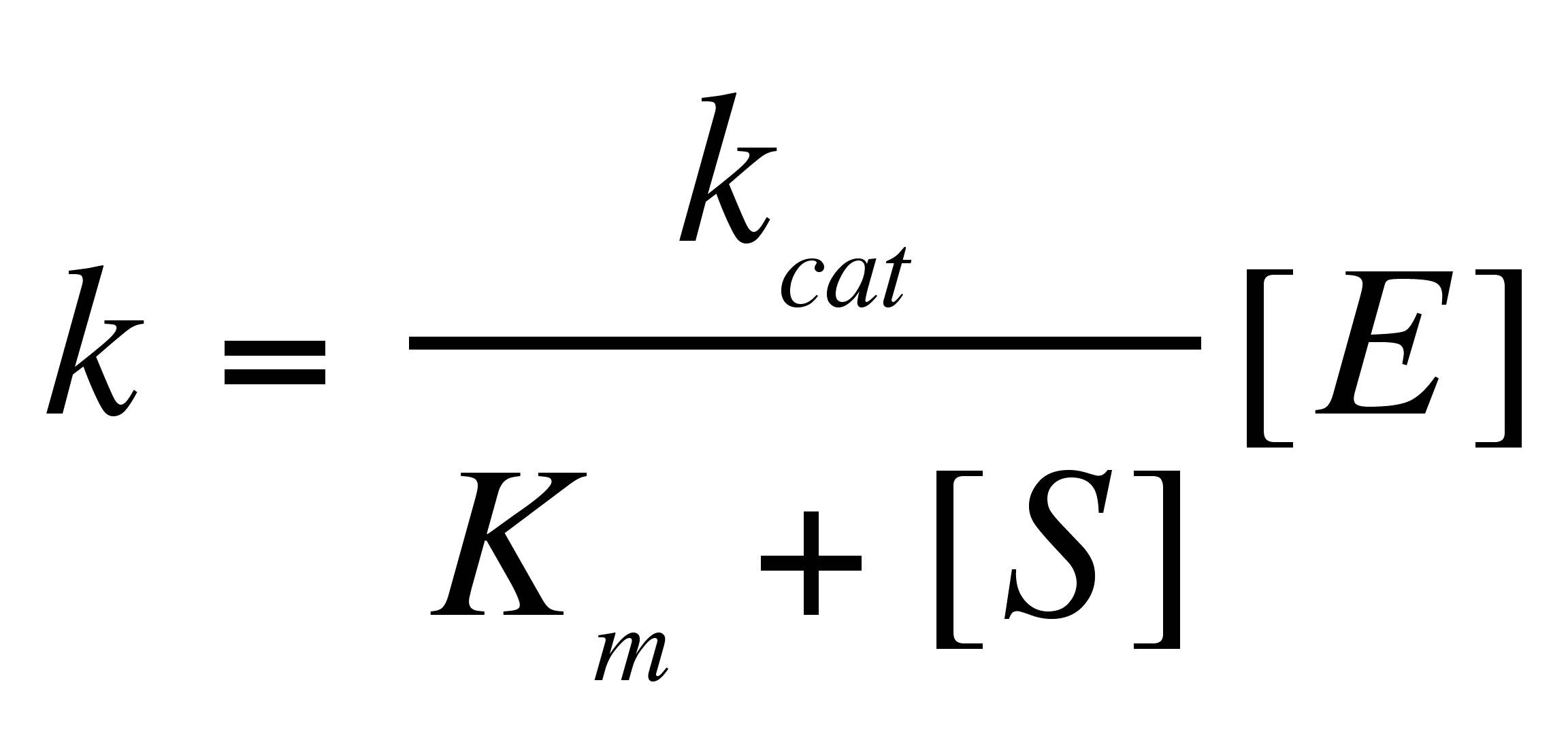 式 (8)
式 (8)
このとき,一連の代謝反応は定常状態にあるとすると基質濃度[S]は一定値をとると考えられるので定数と仮定すると,式 (8) の右辺の[E]以外の項を定数αとおくことで,反応速度定数kは
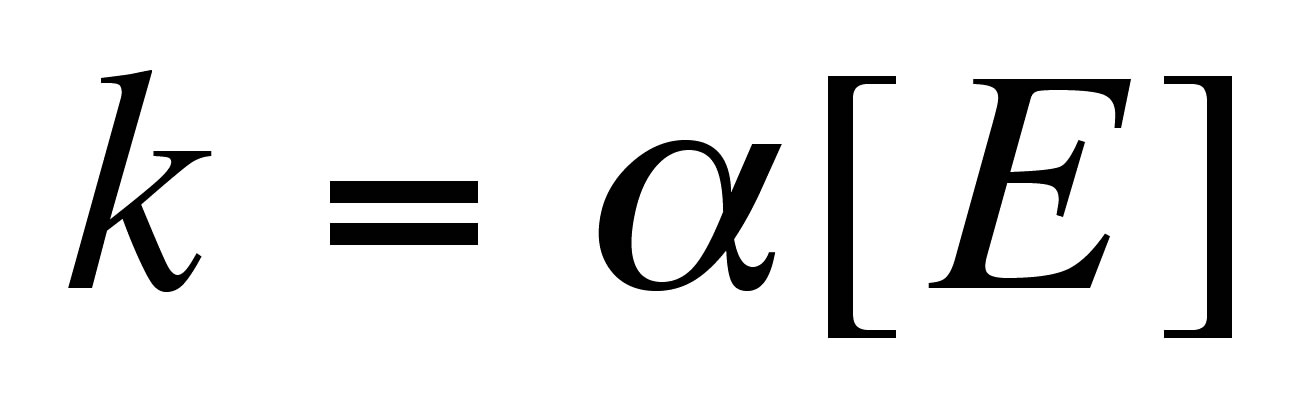 式 (9)
式 (9)
として表される.この値を式 (1) に代入すると,
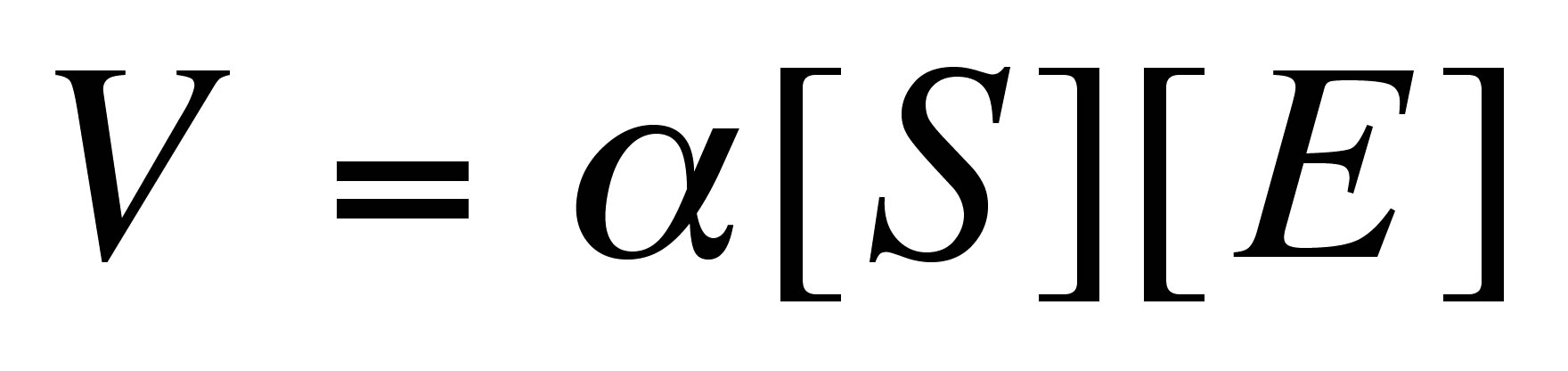 式 (10)
式 (10)
反応速度Vについて解くことができる.なお,Vは反応速度,Vmaは特に質量作用則より導く反応速度,Vmmは特にミカエリスメンテン式より導く反応速度,kは反応速度定数,[S]は基質濃度,[E]は酵素濃度,Vmaxは最大反応速度,Kmはミカエリス定数,kcatは回転数,αは定数をそれぞれ表す.
2.2. マイクロアレイデータとタンパク質存在量との相関解析と回帰分析
本研究では大腸菌を対象生物とした.発現量データは,NCBI GEO (Gene Expression Omnibus; Barrett, et al., 2009) に登録されている大腸菌の全2669サンプル (2010年11月付け) のうち,欠損株やストレス環境下における発現量データを除いた野生株185サンプルを用いた.タンパク質存在量データは,2008年にIshihamaらによってLC-MS/MSを用いて大腸菌の細胞質内におけるタンパク質を990個を同定したデータを使用した (Ishihama, et al., 2008).手法として,タンパク質存在量データに対して発現量データ185個における2群間の相関関係を解析した.次に中心代謝系の解糖系とTCA回路をKEGG Pathway (Kanehisa, et al., 2010) より取得し,各代謝経路に含まれる遺伝子群を抽出した.この抽出した遺伝子群について,マイクロアレイデータとタンパク質存在量データの双方で測定されている遺伝子群を新たに抽出した.抽出した遺伝子群について発現量をタンパク質存在量で割った発現量対タンパク質存在量の比と最小二乗法による単回帰分析を行った.そして,得られた回帰式をもとにマイクロアレイデータをタンパク質存在量データに変換した.ただし,iJR904の902個の遺伝子のうち,Ishihamaらによって定量的に同定された373個のタンパク質は補完対象から除外した.最後に,タンパク質存在量を反応速度との整合性を保つために,大腸菌の乾燥細胞重量を2.8 x 10-13[gDW] (Ishii, et al., 2007) として,質量モル濃度[mmol/gDW]で単位系をそろえた.
2.3. マイクロアレイデータのグループ化と収量の予測
代謝流束解析で収量を計算するモデルとして大腸菌の大規模代謝モデルのひとつであるiJR904 (Reed, et al., 2003) を用いた.iJR904は904個の遺伝子,931個の生体内反応から構成されたモデルである.ただし,本研究においてはiJR904に含まれる遺伝子のうち2遺伝子をアノテーション不足によってマイクロアレイで特定することができなかった.そのため,iJR904で定義される遺伝子のうち902個について解析の対象とした.マイクロアレイデータには,「2.2. マイクロアレイデータとタンパク質存在量との相関解析と回帰分析」の結果より,タンパク質存在量と相関の高いサンプルを使用した.マイクロアレイデータのタンパク質存在量への擬似的な変換には,生体内反応でハウスキーピングな働きをする代謝経路である解糖系とTCA回路より算出した単回帰式と発現量対タンパク質存在量比を用いた (図1).はじめに,902個の遺伝子について発現量を降べき順に並び替えた.次に,マイクロアレイデータのグループ化は,グループ数を決定した後,グループ内に属する遺伝子数を決定した.そして,グループ内の発現量をグループ内の発現量の平均値で均一化した.均一化した発現量をもとに,回帰直線あるいは発現量対タンパク質存在量比で発現量を酵素濃度へ変換し,変換した酵素濃度を反応速度論より導出した方程式へ代入することで各代謝反応の反応速度を計算した.ここで計算した反応速度は,iJR904の反応速度の上限値と下限値として定義した.最後に,目的関数として総菌体量であるバイオマスを設定し代謝流束解析を用いて収量を予測した.なお,代謝流束解析にはBeckerらによって開発されたMATLAB (MathWorks社) 用のツールボックスのひとつであるCOBRA toolbox (Becker, et al., 2007) を利用し,線形計画法の計算には GLPK (GNU Linear Programming Kit; http://www.gnu.org/software/glpk/) を使用した.
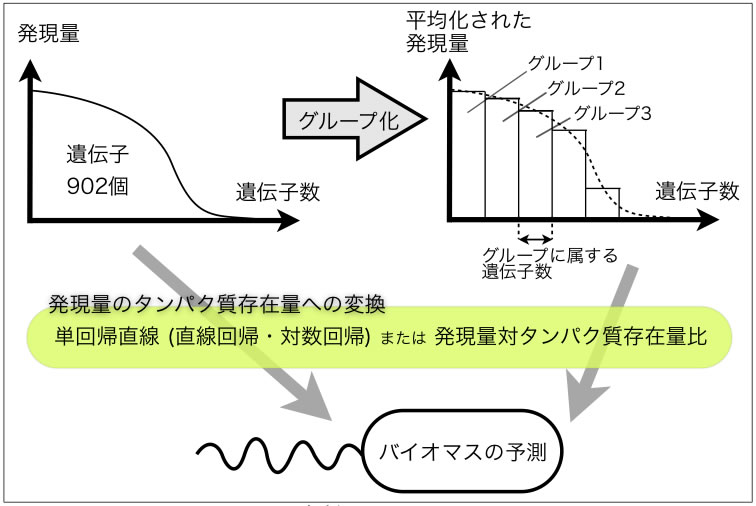 図1: 解析のフローチャート
図1: 解析のフローチャート
マイクロアレイデータのグループ化と収量の予測方法を示した模式図.遺伝子902個の発現量は降べき順に並べた.発現量のグループ化は合計のグループ数を決めた後にグループに含まれる遺伝子数を決定する.次に,グループ内の発現量を一律に平均化し,発現量をタンパク質存在量へと変換する.そして,タンパク質存在量から質量モル濃度へ単位系をそろえ,代謝流束解析と線形計画法よりバイオマスの予測を行った.
3. 結果
3.1. マイクロアレイデータから反応速度の数学的な導出
質量作用則とミカエリスメンテン式より,式 (10)を導いた.このとき,基質濃度[S]は定常状態において定数的に振舞うことが経験的に知られているため,定数であると仮定した.また,αについて,回転数kcat,ミカエリス定数Kmともに定数であるため,αも定数である.以上より,式 (10) におけるα[S]は定数であると考えられる.そして,定数であると仮定したα[S]の定数性については著者が2010年度春学期に行った解析である程度一定の値をとることが示唆された (http://web.sfc.keio.ac.jp/~ike/project/ikegami_2010spring.pdf).この解析は,大腸菌に対して独立に6種類の炭素源を栄養源として与えた実験データ (Liu, et al., 2005) をもとに,発現量データ,反応速度,α[S]について値の分布を調べたところ,α[S]は他の2つにくらべて統計的に有意に値が分散しないことを示した研究である (p<0.0001).したがって,式 (10) で酵素濃度[E]に質量モル濃度に変換した予測値や測定値を代入することによって,反応速度を解くことが可能となった.
3.2. マイクロアレイデータとタンパク質存在量の相関解析
細胞中のmRNA量とタンパク質量が比例関係にあるという仮定を検証し,マイクロアレイデータとタンパク質の相関を解析した.結果,細胞内mRNA量とタンパク質存在量の間には緩やかな相関が見られた (図2).スピアマンの順位相関係数より,最大相関係数は0.65を示し,平均値は0.46,中央値は0.49であった.185個のサンプルで使用されているプラットフォームは7種類存在するが,相関係数0.1以上のサンプル数の約70%がGPL199をプラットフォームとして測定されたマイクロアレイサンプルである.図2の濃い黒色でGPL199のみの場合を示した.プラットフォームごとの平均相関係数ではGPL3154が0.58と最も高く,GPL199の平均相関係数は0.50であった.マイクロアレイサンプルGSM101244が最も高い相関係数を示したが,pH8.7における測定値であったため,同じマイクロアレイシリーズからpH7.0で測定されたGSM101237 (相関係数: 0.60) を本研究においてタンパク質存在量と相関するサンプルとして用いた.なお,マイクロアレイシリーズGSE4511は大腸菌K12株において運動性,酸化ストレス,代謝異化反応に関わる遺伝子群をpHが制御していることを示した実験であり,GSM101244とGSM101237のいずれも野生株,好気条件下で測定されたサンプルであり,プラットフォームにはGPL199を使用しているものである (Maurer, et al., 2005).
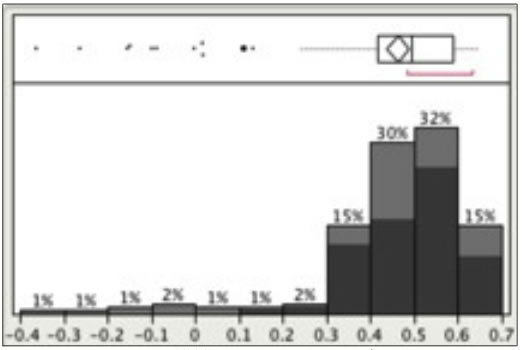 図2: 相関係数のヒストグラム
図2: 相関係数のヒストグラム
発現量データとタンパク質存在量の相関解析の結果.横軸は発現量データとタンパク質存在量の相関係数を表している.灰色は全サンプル数を表し,全サンプルのうちGPL199をプラットフォームとして用いたサンプルを黒色で示した.バーの上に示した数値 (%) はサンプル数の割合である.負の相関を示すデータもあるものの,最も高い相関を示したサンプルGSM101244 (相関係数: 0.65) が含まれるプラットフォームGPL199 (濃い黒色) では,相関係数0.11~0.65,平均相関係数0.50を示した (N=185).
3.3. マイクロアレイデータをタンパク質存在量へ変換
酵素濃度[E]をマイクロアレイデータで補完するために,タンパク質存在量へ変換するすることを試みた.好気条件下でハウスキーピングな代謝経路として恒常的に働く代謝経路である解糖系とTCA回路におけるタンパク質に各データをマッピングし,単回帰直線と発現量対タンパク質存在量の比を求めた.大腸菌K12株の代謝経路では64個の遺伝子が解糖系とTCA回路に関わっており,そのうち約半数の30個の遺伝子がマイクロアレイデータとタンパク質存在量データの双方で測定されたため,この30個の遺伝子を用いた (図3).図3Aはマイクロアレイデータを独立変数としたときの線形回帰 (緑) と対数回帰 (青) を表している.線形回帰のR2値は0.11,対数回帰のR2値は0.37であり.それぞれの回帰式を用いてマイクロアレイデータからタンパク質存在量へと補正した.ただし,グループ数が1個の条件下で,両方の回帰式を用いて反応速度の取りうる上限値を検証したところ,全反応速度の平均値が線形回帰のとき912[mmol/gDW/h],対数回帰のとき1948[mmol/gDW/h]となり,いずれもバイオマスが0.92[gDW/h]を示し,現実的なバイオマスの収量を予測することが出来なかった.野生株 (iJR904) の上限値は一律に1000[mmol/gDW/h]である.そのため,回帰分析の結果を用いると,グループ数を1個に限定しているにも関わらず,野生株と同じくらい,あるいは2倍程度の上限値を設定したことになる.次に,発現量対タンパク質存在量の比を求めた (図3B).発現量対タンパク質存在量比はハウスキーピングに働いている代謝経路においても分散している傾向にあり,本研究では,中央値の1.36を発現量データから擬似的にタンパク質存在量データへと変換するための補正値とした.なお,回帰分析の検証と同様にグループ数を1個に限定したときの全反応速度の上限値の平均値は117[mmol/gDW/h]であった.
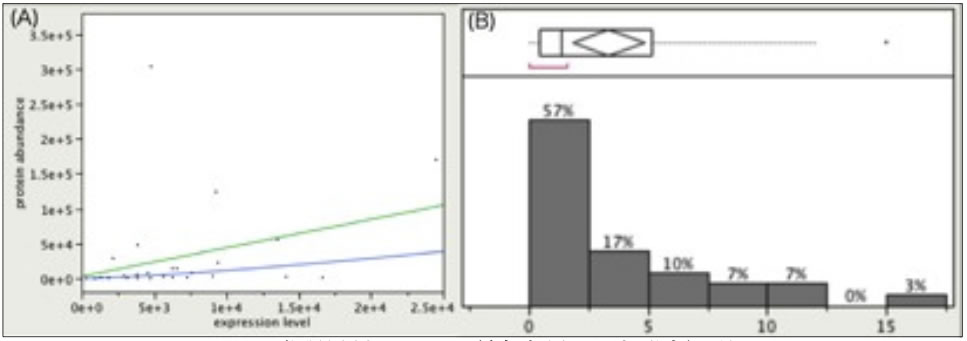 図3: 発現量対タンパク質存在量の回帰分析と比
図3: 発現量対タンパク質存在量の回帰分析と比
(A) 発現量対タンパク質存在量の回帰分析の結果.緑色が線形回帰 (R2=0.11),青色が対数回帰 (R2=0.37) を表している.バイオマスを予測したところ,最も条件の厳しいグループ数が1この時でも最高の収量を示し,推計が過剰であった.(B) 発現量対タンパク質存在量の比の結果.平均は3.33,中央値は1.36を示した.比が広く分布したため中央値を補正値として採用した (N=30).
3.4. マイクロアレイデータのグループ化と収量の予測
発現量のグループ化がバイオマスに与える影響について解析した.図4に示した通りのグループ化を行い,それぞれのバイオマスを予測した (図4).グループ数を増加させ,発現量を平均発現量で均一化したときのバイオマスの変化を示した.グループ数が902個,グループに属する遺伝子数が1個のときが,グループ化の条件が甘く野生株 (iJR904) の条件に近い.このときのバイオマスは,図4中に赤色で塗られている (以下,赤色で塗られたグループ数が902個のモデルを準野生株と呼ぶ).グループ内の遺伝子数が3,5,9,10,15,20のとき,準野生株よりも収量が増す結果となった.特に,遺伝子数が9,10個のときは準野生株よりも約107%収量が増加する.なお,野生株のバイオマスは0.92[gDW/h]であり,準野生株は野生株よりも約21%だけバイオマスの効率が落ちている.グループ内の遺伝子数を増加すると減少傾向を示すものの,単調減少ではなく,上昇と下降を繰り返し,振動しながら減少する様子が計算の結果明らかになった.遺伝子のグループ化とは別に902個の遺伝子すべての遺伝子を (i) 最大の発現量,(ii) 最小の発現量,(iii) 全発現量の平均値,(iv) 全発現量の中央値で均一化し,それぞれバイオマスを予測したところ,最大の発現量で均一化したときバイオマス0.92[gDW/h]を得た.最小値と中央値で均一化したとき線形計画法で最適解に到達することができなかった.他方,平均値で均一化したときバイオマス0.095[gDW/h]となり,グループ内の遺伝子数が902のときバイオマス0.098[gDW/h]とほぼ一致する結果となった.また,バイオマスとグループ数の均衡点として,グループに含まれる遺伝子数が60個のときに,バイオマス0.55[gDW/h]を得る結果となった.
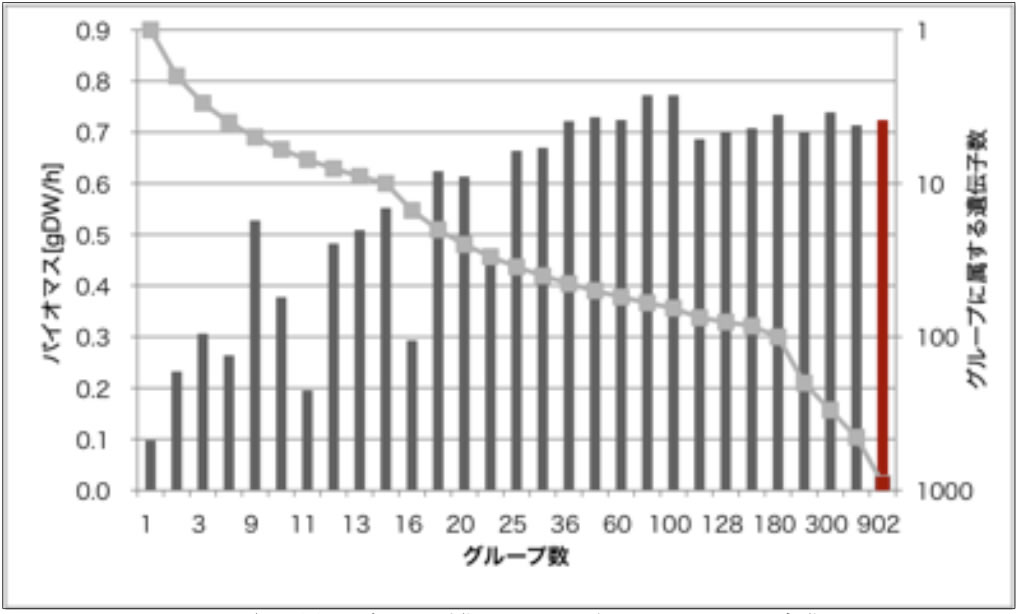 図4: 発現量のグループ化にともなうバイオマスの変化
図4: 発現量のグループ化にともなうバイオマスの変化
バイオマス最大化を代謝流束解析の目的関数とし,収量を予測した.棒グラフはバイオマス現しており,赤色で示した棒と点は,グループ数904個,各グループに含まれる遺伝子数1個,すなわち発現量のグループ化において最も野生株 (iJR904) に近い条件下でのバイオマスを示している.折れ線グラフはグループ数とその条件下でグループに含まれる遺伝子数を表している.
4. 議論
まず,本研究では大腸菌の細胞質内のmRNA発現量とタンパク質存在量は比例すると仮定した.大腸菌の野生株におけるマイクロアレイの網羅的な解析の結果,マイクロアレイデータとタンパク質量データは緩やかな相関関係にあることが確認できた.ただし,マイクロアレイのプラットフォームによって相関関係に偏りが生じているため,相関関係の高かったプラットフォーム3種,あるいは最も高い1種に限定することで,母集団の平均値を高めることができ,今後のバイオマスの予測に用いることができるのではないかと考えられる.その一方で,今回,代謝流束解析に用いたサンプルはプラットフォームGPL199によるデータであるが,iJR904に含まれる遺伝子904個のうち902個だけカバーした.グルコースの比摂取速度10[mmol/gDW/h]のときのフラックス分布を調べたところ,カバーできなかった2個の遺伝子のうち,1つはフラックスをもっていたことを考慮すると,プラットフォームをGPL199の1種に限定すると,1つのフラックに関しては拘束条件を再定義することが難しい可能性がある.
次に,ハウスキーピングな働きをする代謝経路に含まれる遺伝子から,発現量対タンパク質存在量の回帰直線または比を用いて細胞内タンパク質量に変換した.回帰直線は線形回帰 (R2=0.11) と対数回帰 (R2=0.37) を検証してみたところ,いずれの回帰直線もタンパク質存在量を過剰に推計し,代謝流束解析の解の範囲を過大評価した.そのため,グループ数が1個,すなわち全遺伝子の発現量が一律に平均化された条件でも,予測したバイオマスが0.92[gDW/h]となり,それ以降のグループ化もすべてバイオマスが0.92[gDW/h]を示し,グループ化にともなうバイオマスの現象をひとつとして確認することができなかった.そのため,発現量対タンパク質存在量の比を用いて解析を行った.ところが,解糖系とTCA回路に含まれる遺伝子は,常に一定量の発現量をもち,タンパク質存在量も比例関係にあることを想定していたが,予想に反し広い分布を示した.今回は集団の中央値を発現量をタンパク質存在量へ置換する補正値とした.乱暴な補正ではあるが,マイクロアレイの実験誤差とタンパク質定量の実験誤差を考慮すると各反応ごとに発現量対タンパク質存在量比を求め,補正するよりも妥当な補正値となるのではないかと考えた.
マイクロアレイデータから反応速度の上限を決定し,発現量をグループ化した結果,グループ数が902個,グループに属する遺伝子数が1個のとき (準野生株),バイオマスが0.72[gDW/h]であった.COBRAで代謝流束解析を行う場合は各反応速度の取りうる範囲を有限値1000[mmol/gDW/h]に設定してあり,野生株としてバイオマス0.92[gDW/h]が予測される.今回,使用したマイクロアレイのサンプルは大腸菌K12株であるため,本来であれば有限値1000[mmol/gDW/h]の条件下,すなわち代謝モデルにおける野生株のバイオマスと同等の収量が望まれる.しかし,結果としてこのような差異が生じた原因として,交換代謝流における比摂取速度と発現量からタンパク質存在量への補正値の再検討が必要である.今回の解析で反応速度は[mmol/gDW/h],各種濃度は[mmol/gDW]に変換し,一貫性を持たせているため,単位系の間違いによる差異とは考えられにくい.そして,全発現量を最小の発現量で均一化したときと,全発現量の平均値で均一化したときに解を求めることができなかったが,それは野生株におけるフラックスが発現量が弱いために遮断されたからであると考えられる.
また,収量とグループ数の均衡点について,バイオマス0.55[gDW/h]をグループに含まれる遺伝子数が60個,グループ数が約15個のときに得る.グループ数が約15個ということは,代謝関連遺伝子904個を約15種類の擬似的な転写因子によって制御することになる.ただし,準野生株のバイオマスを基準としてバイオマスの5%,10%,15%減少をひとつの閾値としてみなすと,バイオマスがそれぞれ0.68[gDW/h],0.65[gDW/h],0.61[gDW/h]に相当する.そして,グループに含まれる遺伝子数で見ると5%減少のとき25遺伝子,10%と15%減少のとき約50遺伝子に相当する.Gilらは206個の遺伝子を最小ゲノムを構成する遺伝子候補として提唱しているが (Gil, et al., 2004),今回の解析結果を適応すると約4個から9個の転写因子によって,準野生株におけるバイオマスの85%から95%を保証することができるのではないかと考える.また,同様に大腸菌K12株に対して適応すると約86個から172個の転写因子によって準野生株におけるバイオマスの85%から95%を保証することができると考える.事実,RegulonDBでは,大腸菌K12株における転写因子数を175個と定義づけており (Gama-Castro, et al., 2011),準野生株におけるバイオマスの保障性は,今回使用した論理モデルの確からしさを表す結果となった.
5. 展望
バイオマスの予測には,より確からしいタンパク質存在量データが不可欠である.今回の解析では,発現量対タンパク質存在量の回帰分析や比によってマイクロアレイデータをタンパク質存在量データへ補完したが,単回帰直線におけるR2値の低さ,比の分散具合など不確定な条件がめだった.そこで,データは限られているもののIshiiらによって測定された中心代謝系に関わる発現量,タンパク質量,代謝物量のデータ (Ishii, et al., 2007) を用いて改めて発現量対タンパク質存在量の回帰分析や比を求め,その妥当性を検証しバイオマスを予測する必要があると考えている.
今回の解析では,発現量データにもとづいて各反応速度の取りうる上限値を再設定し,代謝流束解析からバイオマスを定量的で予測することはできた.ただし,すべての代謝フラックスが上限値に達しているかは確認していない.むしろ,バイオマスを定義づける物質濃度を代数的に計算し,マイクロアレイデータから直接的にバイオマスを計算し,バイオマスを再計算する必要があると考えている.また,一定量のバイオマスを保証した上で,バイオマスとグループ数の均衡点,バイオマスの保障性について解析することができたものの,バイオマスの予測と実際の細胞増殖に関する影響と生物学的な実現可能性は今後発展させ,予測結果の妥当性を今後検証していきたい.そして,将来的な展望として,転写因子を考慮した最小ゲノムの遺伝子セットのデザインが可能になると期待している.
参考文献
Arakawa,K. et al. (2006) GEM System: automatic prototyping of cell-wide metabolic pathway models from genomes. BMC Bioinformatics, 7, 168.
Barrett,T. et al. (2009) NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res., 37, D885-890.
Becker,S.A. et al. (2007) Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox. Nat Protoc, 2, 727-738.
Edwards,J.S. and Palsson,B.O. (1998) How will bioinformatics influence metabolic engineering? Biotechnol. Bioeng., 58, 162-169.
Edwards,J.S. and Palsson,B.O. (2000) Metabolic flux balance analysis and the in silico analysis of Escherichia coli K-12 gene deletions. BMC Bioinformatics, 1, 1.
Ellis,T.W. et al. (2009) Diversity-based, model-guided construction of synthetic gene networks with predicted functions. Nat. Biotechnol., 27, 465-471.
Elowitz,M.B. and Leibler,S. (2000) A synthetic oscillatory network of transcriptional regulators. Nature, 403, 335-338.
Fong,S.S. and Palsson,B.O. (2004) Metabolic gene-deletion strains of Escherichia coli evolve to computationally predicted growth phenotypes. Nat. Genet., 36, 1056-1058.
Gama-Castro,S et al. (2011) RegulonDB version 7.0: transcriptional regulation of Escherichia coli K-12 integrated within genetic sensory response units (Gensor Units). Nucleic Acids Res., 39, D98-105.
Gibson,D.G. et al. (2010) Creation of a Bacterial Cell Controlled by a Chemically Synthesized Genome. Science, 329, 52-56.
Gil,R. et al. (2004) Determination of the core of a minimal bacterial gene set. Microbiol. Mol. Biol. Rev., 68, 518-537.
Ishihama,Y. et al. (2008) Protein abundance profiling of the Escherichia coli cytosol. BMC genomics, 9, 102.
Ishii,N. et al. (2007) Multiple high-throughput analyses monitor the response of E. coli to perturbations. Science, 316, 593-597.
Itaya,M. et al. (2008) Bottom-up genome assembly using the Bacillus subtilis genome vector. Nat Methods, 5, 41-43.
Kanehisa,M. et al. (2010) KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res., 38, D355-360.
Koonin,E.V. (2003) Comparative genomics, minimal gene-sets and the last universal common ancestor. Nat. Rev. Microbiol., 1, 127-136.
Liu,M. et al. (2005) Global transcriptional programs reveal a carbon source foraging strategy by Escherichia coli. J. Biol. Chem., 280, 15921-15927.
Maurer,L.M. et al. (2005) pH regulates genes for flagellar motility, catabolism, and oxidative stress in Escherichia coli K-12. J. Bacteriol., 187, 304-319.
Moya,A. et al. (2009) Toward minimal bacterial cells: evolution vs. design. FEMS Microbiol. Rev., 33, 225-235.
Purnick,P.E. and Weiss,R. (2009) The second wave of synthetic biology: from modules to systems. Nat. Rev. Mol. Cell Biol., 10, 410-422.
Reed,J.L. and Palsson,B.O. (2003) Thirteen years of building constraint-based in silico models of Escherichia coli. J. Bacteriol., 185, 2692-2699.
Reed,J.L. et al. (2003) An expanded genome-scale model of Escherichia coli K-12 (iJR904 GSM/GPR). Genome Biol., 4, R54.
Reed,J.L. et al. (2006) Towards multidimensional genome annotation. Nat. Rev. Genet., 7, 130-141.