
政策・メディア研究科 玉木 聡志
Phosphorylation is one of the most important post-translational modification. Phosphorylation is involved in multiple biological process such as DNA damage and repair, transcrip- tion regulation, and metabolism. In human proteome, three types of major residue in the substrate; serine(S), threonine(T), and tyrosine (Y) is phosphorylated by their responsible kinase. Streaming motion of phosphorylation constructs an signal transaction network of the living cell.
Resent achievements in the area of mass spectrometry based proteomics accomplished in determining the phosphorylated residue of the protein in a massive scale. On the other hand, the global picture of signal network driven by phosphorylation still remains unclear, due to the lack of information of kinase-substrate pairs. To work out with this problem, computational approaches has been applied. While various numbers of computational predictors such as Scansite, NetworKIN, PPRED are available, there still remains a interruption in constructing a kinase specific predictor in a global mass scale.
To this end, we have created a kinase specific predictor of 269 human kinase, with high accuracy. By combining the in vitro phosphoproteimics approche with machine learning techniques, we were able to createda phosphoredisue predictor with a resolution of single kinase. High accuracy predictor used in the method is easily applicative in searching the unknown phosphorylation sites of the kinase. In this paper, we will first discuss about creating the non kinase specific classifier and discuss the potential of expanding it to kinase specific classifier.
In this study, we have created 2types of predictor. SVM based predictor using sequence features around the phosphorylated residue, and deep neural network using 3d atom postions around the phosphorylated residue. In constructing the svm phosphorylation prediction model, we have basically followed the method proposed in the PPRED paper. The overall workflow is shown in Figure1. The optimal C and gamma used for SVM training was additionally performed.
Sugiyama et al, has obtained a large-scale information of human kinase-substrate pairs in a macro scale. By reacting 347 recombinant kinase originated from human in vitro and measuring the phosphorylated residue responsible for that kinase [personal communication, an article under submission]. Using the PSSM created by PSI-BLAST, matrix of 5 residue before and after S/T/Y was extracted, and used for the SVM training. The positive and negative dataset was trained, using libSVM software. For the number of SVM training, the ratio between positive and negative critically affects the prediction accuracy. Since the number of negative dataset is comparatively larger than the positive dataset, we have randomly reduced the data in the negative dataset until the ratio of positive : negative achieved 1:1. In the process in creating the model, grid search was performed by using the grid.py in the LIBSVM Tools to obtain optimum C, and gamma in classification. For each phosphorylated residue, C and gamma was determined.
To assure that the predicted phosphoresidue is not biased on our experimental process, we have obtained the positions of phosphorylated residue database, independent from our experiment . We have used these phosphoresidue locations for validation of the classifier. From UniProt KB, position of the phosphorylated residue was determined by referring the ’Modified residue’ from the Features annotation.
All the human protein data that had Phosphosite plus and PDB was collected. The position of phosphorylated residue was determined using Phosphosite Plus. From the PDB entry, we have counted the composition of residues per 1 angstrom. The count of each residue was used as the feature for deep neural net training. Software caffe was selected for training. GPGPU calculation was done using the amazon ec2 GPU machine.
The ROC curve of the non-kinase specific predictor from the result of three-fold cross validation before and after the grid parameter search is shown. ROC curve in Figure1 is created by following the method shown in the paper of PPRED.
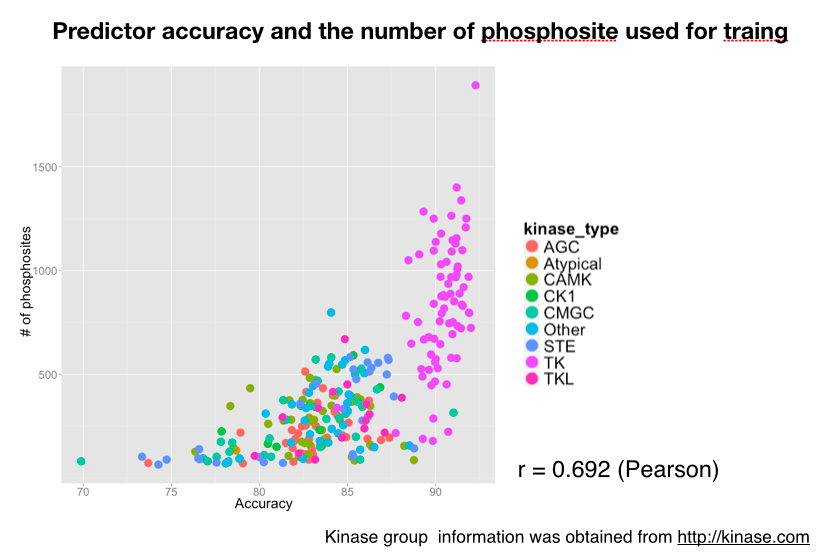
The accuracy histogram of the created 269 kinase-specific classifier is shown in Figure 3. To validate the relationships between the accuracy of each kinase predictor and the phosphopeptide used for training, scatter plot of the accuracy of the predictor is shown below.
We have constructed a DNN based predictor of kinase PKACA and PKCAand compared the accuracy of predictor with the svm using the same features. The DNN precitor showed higher prediction rate compared to the svm predictor
| Predictor | Accuracy |
|---|---|
| DNN | 62.2% |
| SVM | 56.6% |
In this work, we have created 2 types of predictor. First is the 269 kinase specific predictor, and 230/269 predictors showed an accuracy over 80%. This accuracy and resolution of the predictor outperforms the existing predictors. As we can observe from Figure 2.2, predictors belonging to tyrosine kinase group showed an especially high performance. It is known that the tyrosine kinase substrates does not have an well-defined motif, and is difficult to predict the kinase substrate pairs with the existing prediction method based on sequence search. This aspect is a clear advantage using our prediction method.
The Second predictor is the Deep neural network predictor. using the 3D atom positons around the phosphorylated residue, we were able to develop a predictor of the phoshorylated positions in hight accuracy.

Large scale phosphoproteomics approach enabled us to create un kinase specific predictor in massive scale and resolution. Since the accuracy of the kinase specific predictor and the numbers of the training dataset used for constructing the model showed a loose correlation. Further technical improvement in the field of phosphoproteomics, will expand the quality of the predictor.
I am deeply grateful to Kazuharu Arakawa, for being a grate adviser and opened the door to the world of science. From Shinya Murata my second adviser, I learned a way of living a flexible life. Rintaro Saito trained me in the field of statistics and transcriptomics. Prof Akihiko Kanaya, my academic supervisor of the masters course in Nara Institute of Science and Technology, inspired my vision of living as a biological scientist. This work was supported by the Mori Grants 2015. Finally, I would like to Thank Prof Masaru Tomita for being my academic supervisor of undergrad and doctors course